- Kunstnerisk fremstilling
- Figur1:Hieronymous Bosch, De jordiske glædernes have, et forstørret udsnit af det højre panel af det indre afsnit (Helvede). Det groteske øre har en struktur, der minder om en PauS (pil).
- Første medicinske beskrivelser
- Figur2:Klassisk fremstilling af PAuS (pil).
- Figur3:Variant af PAuS på den aurikulære helix (pil).
- PAuS-associerede syndromer
- Melnick-Fraser syndrom
- Beckwith-Wiedemann syndrom
- Lachiewicz-Sibley syndrom
- Enogtyvende århundrede
Kunstnerisk fremstilling
Hieronymus Bosh (ca. 1450-1516) var en hollandsk kunstner, hvis mest berømte maleri er “De jordiske glædernes have”. Maleriet illustrerer udvendige og indvendige paneler . Det ydre panel ligner verdens skabelse, mens det indre består af tre paneler, der forestiller henholdsvis Edens Have (venstre panel), Den menneskelige verden (midterste panel) og Dommedag eller Helvede (højre panel). Et lille udsnit af det højre panel illustrerer et par ører, hvorpå der ses en struktur, der ligner en PAuS (Figur 1) .

Figur1:Hieronymous Bosch, De jordiske glædernes have, et forstørret udsnit af det højre panel af det indre afsnit (Helvede). Det groteske øre har en struktur, der minder om en PauS (pil).
PAuS, preauricular sinus
Første medicinske beskrivelser
PAuS blev første gang beskrevet i 1864 af C. F. Heusinger, da han beskrev fundene hos en patient, der var karakteristisk for brachio-oto-renal (BOR) syndromet . Han beskrev sit fund i detaljer og henviste også til flere allerede beskrevne fænomener, der syntes løsrevet fra hinanden, såsom det indledende arbejde af Dzondi, som beskrev og definerede medfødte tracheale fistler . Det er vigtigt at bemærke, at den af Heusinger beskrevne auricula-enhed, selv om den er morfologisk identisk, er en selvstændig enhed i forhold til de cervikale og andre brachiale fistler, der var beskrevet før hans observationer .
I forlængelse af Heusingers beskrivelse beskrev Rudolph Virchow (1821-1902) også strukturen i 1864, selv om han i sin artikel blot anførte: “Jeg kender også en patient som denne” . Virchow var dog den første til at postulere, at PAuS er et resultat af en defekt i den embryologiske fusion af svælgbuerne, hvilket var et udsagn, der blev bredt modsagt på daværende tidspunkt (Figur 2).
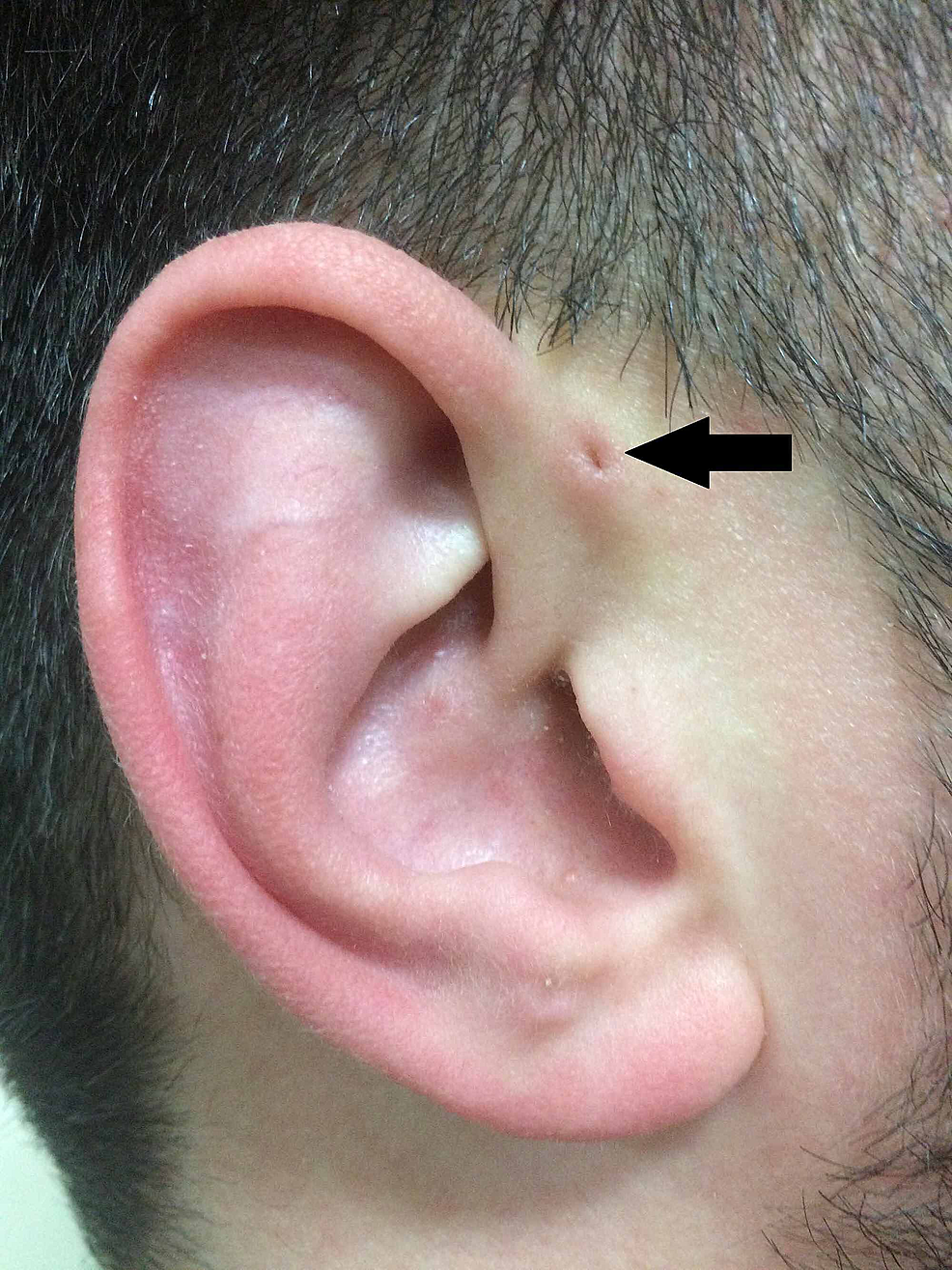
Figur2:Klassisk fremstilling af PAuS (pil).
PAuS, preauricular sinus
Sir James Paget offentliggjorde også om emnet i 1878 og beskrev patienter med sådanne fistler, der ikke kun var til stede på ørerne, men også på halsen . På baggrund af beskrivelsen af sine patienter opfandt han udtrykket oto-branchial fistel (figur 3). Sir James Paget tilpassede i vid udstrækning synspunkterne i Virchows teori om oprindelsen af PAuS.
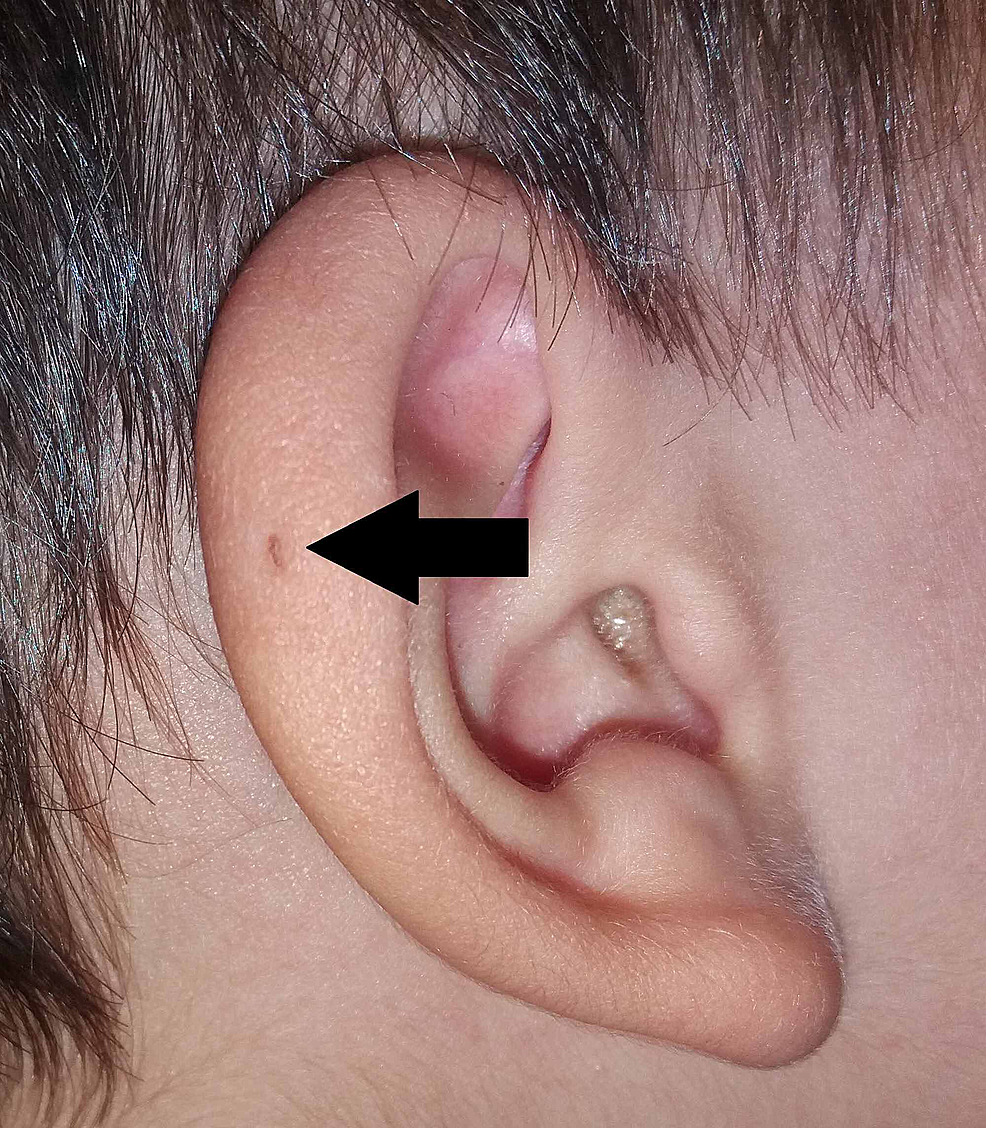
Figur3:Variant af PAuS på den aurikulære helix (pil).
PAuS, preauricular sinus
I første del af det 20. århundrede blev PAuS kendt som “naturlige ørehuller” og “fistula auris congenita”, og flere forfattere har publiceret om emnet, herunder om dets arvegangsmønster . Fænomenet og dets transgenerationelle arv gav mulighed for en omfattende række undersøgelser og teorier om dets årsag, der i vid udstrækning konvergerede mod Virchows oprindelige udsagn .
Et af de vigtigste bidrag fra denne tidsperiode blev i 1955 af Fourman og Fourman, der undersøgte arven af PAuS og konstruerede et genealogisk træ, med fokus på arven af strukturen . De konkluderede, at halvdelen af søskende i den berørte familie har strukturen og postulerede således, at sinus arves som et dominerende træk, men med “ufuldstændig penetrering” på grund af, at den springer over i generationer eller individer .
På trods af denne indledende interesse blev der imidlertid kun offentliggjort få eller ingen artikler efter slutningen af 1940’erne, indtil en lille genopblussen i de senere år, hvor strukturen genopstår under et nyt navn – PAuS. Siden da er der sjældent blevet forsket i strukturen, og mekanismerne for dens transgenerationelle nedarvning er stadig i vid udstrækning glemt i litteraturen fra 1930’erne og 1940’erne.
PAuS-associerede syndromer
Melnick-Fraser syndrom
Syndromet blev første gang beskrevet i 1864 af Heusinger . I 1975 beskrev Melnick et al. imidlertid en serie af patienter med det, og i 1980 beskrev Fraser et al. endnu en serie af tilfælde med syndromets kendetegn på en specialskole for døve i en specialskole for døve . Tilstanden anses for at være relativt sjælden, idet 250 nye tilfælde af Melnick-Fraser blev diagnosticeret i Japan i 2014 . Først troede man, at det var en variant af BOR-syndromet, men senere udvidede Melnick syndromet til en gruppe af næsten identiske tilstande, der afhænger af tilstedeværelsen eller fraværet af forskellige kendetegn . Udtrykket Melnick-Frasers syndrom blev senere opfundet for at forene de separate syndromers karakteristika og for helt at adskille det fra Frasers syndrom, isolerede urogenitale misdannelser, og Frasier-Lynch syndrom, familiær kolorektal polypose, som BOR ret ofte var blevet forvekslet med som et udtryk på det tidspunkt. Heusingers bidrag er således desværre blevet bredt glemt.
Beckwith-Wiedemann syndrom
Sygdommen blev oprindeligt beskrevet i 1963 af den amerikanske patolog John Beckwith som en kombination af exomphalos, makroglossia og gigantisme, derfor benævnt exomphalos-macroglossia-gigantisme (EMG) syndrom (Poster: Beckwith, JB: Extreme Cytomegaly of the adrenal Fetal Cortex, Hyperplasia of the Kidneys and Pancreas, and Leydig-Cell Hyperplasia : Another Syndrome?. Annual Meeting of Western Society for Pediatric Research, Los Angeles, CA, 11. november 1963). I 1964 beskrev den tyske børnelæge Hans-Rudolph Wiedemann imidlertid uafhængigt af John Beckwith også flere patienter med de samme kendetegn ved sygdommen, men også med symptomer som binyrebarkhypoplasi . Med tiden blev syndromet døbt Beckwith-Wiedemann-syndromet, og de diagnostiske kriterier blev udvidet til at omfatte PAuS, hyperplastiske nyrer, mikrocefali, neonatal hypoglykæmi og hepatoblastom, der udvikles senere i livet . Mutationer i 11p15, der involverer gener som insulinlignende vækstfaktor 2 (IGF-2), cyclinafhængig kinaseinhibitor 1C (CDKN1C), H19 og kaliumspændingsstyret kanal underfamilie Q-medlem 1 overlappende transkript 1 (KCNQ1OT1), er blevet konstateret hos sådanne patienter . Arvemekanismen er stadig udefineret, da den nøjagtige transgenerationelle arv hos flere patienter med denne tilstand ikke kunne bestemmes, hvilket betegner den som en sporadisk recessiv defekt. Beckwith-Wiedemanns syndrom og dets kendetegn er særligt vigtige at skelne mellem patienter, der er undfanget ved in vitro-befrugtning, da incidensen i denne population er meget højere . Beckwith-Wiedemann betragtes som en sjælden tilstand med en forekomst på 1 pr. 13 700 fødte børn, hvilket svarer til i alt ca. 300 børn født med Beckwith-Wiedemann i USA årligt .
Lachiewicz-Sibley syndrom
Dette syndrom blev først beskrevet i 1985 af Lachiewicz et al. og er et af de mest sjældne syndromer, der nogensinde er beskrevet . Syndromet ligner meget Melnick-Fraser, selv om kun PAuS og hypoplastiske nyrer med tidligt indsættende proteinuri blev fundet hos efterkommere af britiske og irske immigranter, der bosatte sig i Ohio i 1800-tallet og senere Nebraska. På tidspunktet for den oprindelige undersøgelse havde 12 medlemmer af 130 levende slægtninge ud af 130 slægtninge PAuS og hypoplastiske nyrer, 10 havde kun hypoplastiske nyrer, og 3 havde kun PAuS . Selv om det nøjagtige mutationslokus fortsat er ukendt, nedarves tilstanden autosomalt dominerende.
Enogtyvende århundrede
I dag er PAuS, selv om det stadig er relativt lidt udforsket, en almindelig klinisk enhed med veldefinerede kliniske strategier for behandling. Nogle undersøgelser har identificeret en genetisk sammenhæng mellem PAuS og locus 8q11.1-13.1; resultaterne er dog ikke blevet reproduceret bredt . Bortset fra denne enkelte undersøgelse har ingen andre undersøgelser fokuseret på den genetiske årsag til PauS eller har associeret den med de førnævnte genetiske syndromer, med genealogiske undersøgelser er få og langt imellem .
Det kliniske forløb er velbeskrevet, og behandlingsstrategier, herunder overvejende kirurgisk excision, er veletableret i moderne otorhinolaryngologi og hoved- og halskirurgi . I dag gennemgår patienter med PAuS, der ikke er forbundet med et genetisk syndrom, en sikker interventionsproces med få beskrevne komplikationer, mens patienter med genetiske syndromer gennemgår de samme kurative modaliteter, og excisionen af PauS påvirker ikke deres samlede kliniske forløb .