- Künstlerische Darstellung
- Abbildung1:Hieronymous Bosch, Garten der Lüste, ein vergrößerter Ausschnitt der rechten Tafel des inneren Teils (Hölle). Das groteske Ohr hat eine Struktur, die an ein PauS erinnert (Pfeil).
- Erste medizinische Beschreibungen
- Abbildung2:Klassische Darstellung des PAuS (Pfeil).
- Abbildung3:Variante des PAuS an der Ohrmuschelhelix (Pfeil).
- PAuS-assoziierte Syndrome
- Melnick-Fraser-Syndrom
- Beckwith-Wiedemann-Syndrom
- Lachiewicz-Sibley-Syndrom
- Einundzwanzigstes Jahrhundert
Künstlerische Darstellung
Hieronymus Bosh (ca. 1450-1516) war ein niederländischer Künstler, dessen bekanntestes Gemälde „Der Garten der Lüste“ ist. Das Gemälde zeigt eine Außen- und eine Innentafel. Die äußere Tafel stellt die Erschaffung der Welt dar, während das Innere aus drei Tafeln besteht, die jeweils den Garten Eden (linke Tafel), die Welt der Menschen (mittlere Tafel) und den Tag des Jüngsten Gerichts oder die Hölle (rechte Tafel) darstellen. Ein kleiner Ausschnitt der rechten Tafel zeigt ein Paar Ohren, auf denen eine Struktur zu sehen ist, die einer PAuS ähnelt (Abbildung 1).

Abbildung1:Hieronymous Bosch, Garten der Lüste, ein vergrößerter Ausschnitt der rechten Tafel des inneren Teils (Hölle). Das groteske Ohr hat eine Struktur, die an ein PauS erinnert (Pfeil).
PAuS, präaurikulärer Sinus
Erste medizinische Beschreibungen
Das PAuS wurde erstmals 1864 von C. F. Heusinger beschrieben, als er den Befund bei einem Patienten beschrieb, der für das brachio-oto-renale (BOR) Syndrom charakteristisch war. Er beschrieb seinen Befund ausführlich und verwies auch auf mehrere bereits beschriebene Phänomene, die voneinander losgelöst zu sein schienen, wie die Vorarbeiten von Dzondi, der angeborene Trachealfisteln beschrieb und definierte. Es ist wichtig festzuhalten, dass die von Heusinger beschriebene Auricula-Entität, obwohl sie morphologisch identisch ist, eine von den zervikalen und anderen brachialen Fisteln, die vor seinen Beobachtungen beschrieben wurden, getrennte Entität ist.
Nach Heusingers Beschreibung beschrieb auch Rudolph Virchow (1821-1902) 1864 die Struktur, obwohl er in seinem Artikel lediglich feststellte „Ich kenne auch einen solchen Patienten“ . Virchow war jedoch der erste, der postulierte, dass das PAuS eine Folge eines Defekts in der embryologischen Verschmelzung der Rachenbögen ist, eine Aussage, die zu dieser Zeit weithin widersprochen wurde (Abbildung 2).
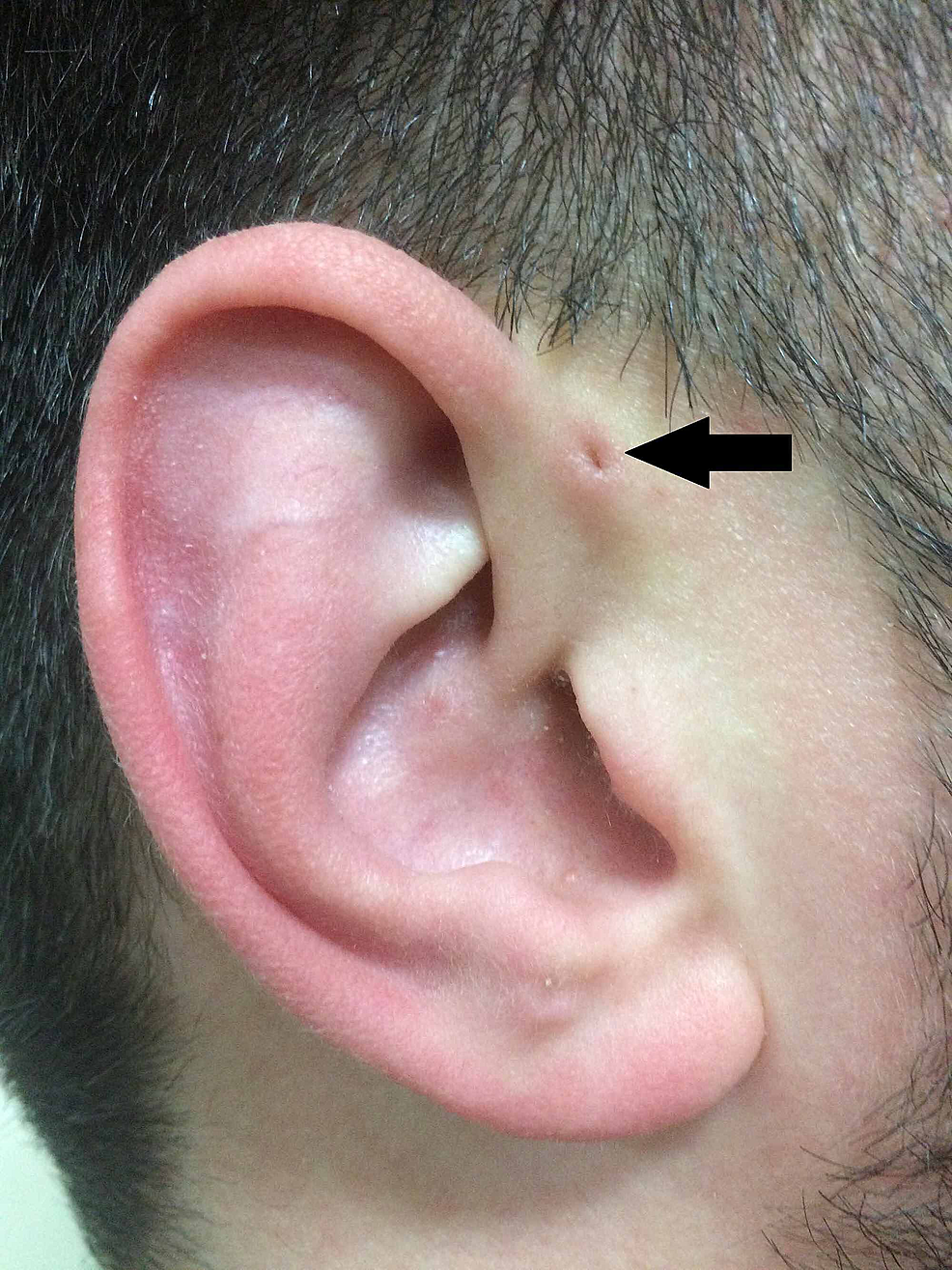
Abbildung2:Klassische Darstellung des PAuS (Pfeil).
PAuS, präaurikulärer Sinus
Sir James Paget veröffentlichte 1878 ebenfalls zu diesem Thema und beschrieb Patienten mit solchen Fisteln, die nicht nur an den Ohren, sondern auch am Hals vorhanden waren. Auf der Grundlage der Beschreibung seiner Patienten prägte er den Begriff der oto-branchialen Fistel (Abbildung 3). Sir James Paget übernahm weitgehend die Ansichten von Virchows Theorie über den Ursprung des PAuS.
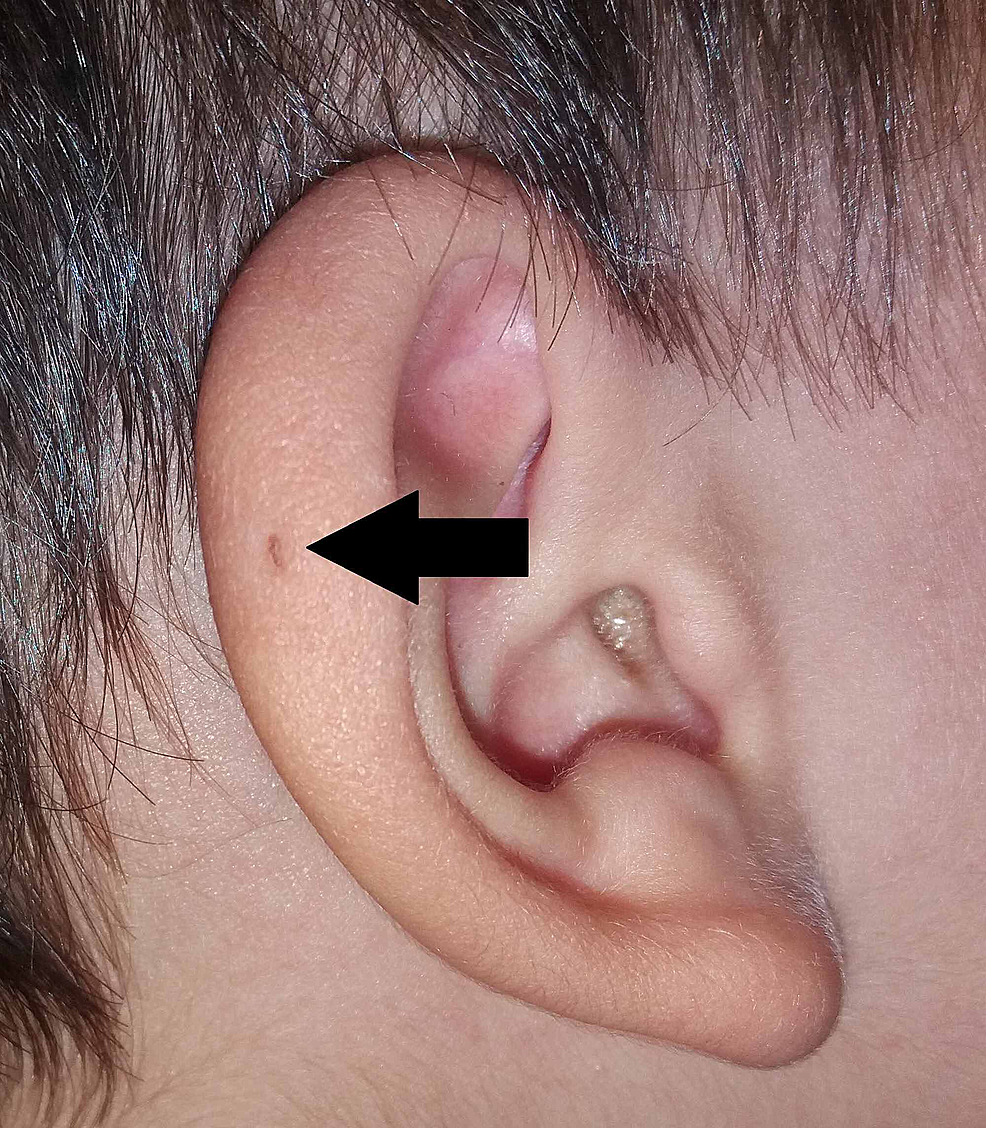
Abbildung3:Variante des PAuS an der Ohrmuschelhelix (Pfeil).
PAuS, präaurikulärer Sinus
In der ersten Hälfte des 20. Jahrhunderts wurde das PAuS als „natürliche Ohrlöcher“ und „Fistula auris congenita“ bekannt, und mehrere Autoren veröffentlichten zu diesem Thema, einschließlich seines Vererbungsmusters. Das Phänomen und seine transgenerationale Vererbung ermöglichten eine Vielzahl von Studien und Theorien zu seiner Ursache, die sich weitgehend mit Virchows ursprünglichen Aussagen deckten.
Einer der wichtigsten Beiträge aus dieser Zeit stammt aus dem Jahr 1955 von Fourman und Fourman, die die Vererbung von PAuS untersuchten und einen Stammbaum mit Schwerpunkt auf der Vererbung der Struktur erstellten. Sie kamen zu dem Schluss, dass die Hälfte der Geschwister der betroffenen Familie die Struktur haben, und postulierten daher, dass der Sinus als dominantes Merkmal vererbt wird, jedoch mit „unvollständiger Penetranz“, da er Generationen oder Individuen überspringt.
Trotz dieses anfänglichen Interesses wurden jedoch nach den späten 1940er Jahren nur wenige bis gar keine Artikel veröffentlicht, bis zu einem kleinen Wiederaufleben in späteren Jahren, in denen die Struktur unter einem neuen Namen – PAuS – wieder auftauchte. Seitdem ist die Struktur nur noch selten erforscht worden, und die Mechanismen ihrer transgenerationalen Vererbung sind in der Literatur der 1930er und 1940er Jahre weitgehend in Vergessenheit geraten.
PAuS-assoziierte Syndrome
Melnick-Fraser-Syndrom
Das Syndrom wurde erstmals 1864 von Heusinger beschrieben. Im Jahr 1975 beschrieben Melnick et al. jedoch eine Reihe von Patienten mit diesem Syndrom, und 1980 beschrieben Fraser et al. eine weitere Reihe von Fällen mit den Merkmalen des Syndroms in einer Spezialschule für Gehörlose. Die Erkrankung gilt als relativ selten, wobei 2014 in Japan 250 neue Fälle von Melnick-Fraser diagnostiziert wurden. Zunächst wurde angenommen, dass es sich um eine Variante des BOR-Syndroms handelt, doch später erweiterte Melnick das Syndrom zu einer Gruppe nahezu identischer Erkrankungen, die vom Vorhandensein oder Fehlen verschiedener Merkmale abhängen. Der Begriff Melnick-Fraser-Syndrom wurde später geprägt, um die Merkmale der einzelnen Syndrome zu vereinheitlichen und es vollständig vom Fraser-Syndrom, isolierten urogenitalen Fehlbildungen, und dem Frasier-Lynch-Syndrom, familiärer kolorektaler Polyposis, abzugrenzen, mit denen BOR zu diesem Zeitpunkt häufig verwechselt wurde. So ist Heusingers Beitrag leider weitgehend in Vergessenheit geraten.
Beckwith-Wiedemann-Syndrom
Das Krankheitsbild wurde ursprünglich 1963 von dem amerikanischen Pathologen John Beckwith als eine Kombination von Exomphalos, Makroglossie und Gigantismus beschrieben und daher als Exomphalos-Makroglossie-Gigantismus (EMG)-Syndrom bezeichnet (Poster: Beckwith, JB: Extreme Zytomegalie der fetalen Nebennierenrinde, Hyperplasie der Nieren und des Pankreas und Leydig-Zell-Hyperplasie : Ein anderes Syndrom? Jahrestagung der Western Society for Pediatric Research, Los Angeles, CA, 11. November 1963). Unabhängig von John Beckwith beschrieb der deutsche Kinderarzt Hans-Rudolph Wiedemann 1964 ebenfalls mehrere Patienten mit den gleichen Merkmalen der Krankheit, aber auch mit Symptomen wie einer Nebennierenhyperplasie. Im Laufe der Zeit wurde das Syndrom als Beckwith-Wiedemann-Syndrom bezeichnet, und die diagnostischen Kriterien wurden um PAuS, hyperplastische Nieren, Mikrozephalie, neonatale Hypoglykämie und ein sich später im Leben entwickelndes Hepatoblastom erweitert. Bei diesen Patienten wurden Mutationen in 11p15 festgestellt, die Gene wie den insulinähnlichen Wachstumsfaktor 2 (IGF-2), den Cyclin-abhängigen Kinase-Inhibitor 1C (CDKN1C), H19 und das überlappende Transkript 1 (KCNQ1OT1) des spannungsabhängigen Kaliumkanals der Unterfamilie Q, Mitglied 1, betreffen. Der Mechanismus der Vererbung ist noch nicht geklärt, da bei mehreren Patienten mit dieser Erkrankung die genaue transgenerationale Vererbung nicht bestimmt werden konnte, so dass es sich um einen sporadisch rezessiven Defekt handelt. Das Beckwith-Wiedemann-Syndrom und seine Merkmale sind bei Patienten, die durch In-vitro-Fertilisation gezeugt wurden, besonders wichtig, da die Inzidenz in dieser Population viel höher ist. Das Beckwith-Wiedemann-Syndrom gilt als seltene Erkrankung mit einer Inzidenz von 1 pro 13.700 geborenen Kindern, was einer Gesamtzahl von etwa 300 Kindern entspricht, die in den Vereinigten Staaten von Amerika jährlich mit dem Beckwith-Wiedemann-Syndrom geboren werden.
Lachiewicz-Sibley-Syndrom
Dieses Syndrom wurde erstmals 1985 von Lachiewicz et al. beschrieben und ist eines der seltensten jemals beschriebenen Syndrome. Das Syndrom ist dem Melnick-Fraser-Syndrom sehr ähnlich, obwohl bei den Nachkommen britischer und irischer Einwanderer, die sich in den 1800er Jahren in Ohio und später in Nebraska niederließen, nur PAuS und hypoplastische Nieren mit früh einsetzender Proteinurie gefunden wurden. Zum Zeitpunkt der ursprünglichen Studie hatten von 130 lebenden Verwandten 12 Mitglieder PAuS und hypoplastische Nieren, 10 hatten nur hypoplastische Nieren und 3 hatten nur PAuS. Obwohl der genaue Ort der Mutation nicht bekannt ist, wird die Erkrankung autosomal-dominant vererbt.
Einundzwanzigstes Jahrhundert
Heute ist das PAuS, obwohl noch relativ wenig erforscht, eine häufige klinische Entität mit gut definierten klinischen Behandlungsstrategien. In einigen Studien wurde ein genetischer Zusammenhang zwischen PAuS und dem Locus 8q11.1-13.1 festgestellt; die Ergebnisse wurden jedoch nicht umfassend reproduziert. Abgesehen von dieser einzelnen Studie haben sich keine anderen Studien auf die genetischen Ursachen des PauS konzentriert oder es mit den oben genannten genetischen Syndromen in Verbindung gebracht, und genealogische Studien sind selten.
Der klinische Verlauf ist gut beschrieben, und die Behandlungsstrategien, einschließlich der überwiegend chirurgischen Entfernung, sind in der modernen HNO-Heilkunde und Kopf- und Halschirurgie gut etabliert. Heutzutage werden Patienten mit PAuS, die nicht mit einem genetischen Syndrom assoziiert sind, einem sicheren Interventionsprozess mit wenigen beschriebenen Komplikationen unterzogen, während Patienten mit genetischen Syndromen denselben kurativen Modalitäten unterzogen werden und die Entfernung des PauS keinen Einfluss auf ihren gesamten klinischen Verlauf hat.