- Representación artística
- Figura1:El Bosco, El jardín de las delicias terrenales, una sección ampliada del panel derecho de la sección interior (El infierno). La oreja grotesca tiene una estructura que recuerda a un PauS (flecha).
- Primeras descripciones médicas
- Figura2:Representación clásica del PAuS (flecha).
- Figura3:Variante de PAuS en el hélix auricular (flecha).
- Síndromes asociados al PAuS
- Síndrome de Melnick-Fraser
- Síndrome de Beckwith-Wiedemann
- Síndrome de Lachiewicz-Sibley
- Siglo XXI
Representación artística
Hieronymus Bosh (c. 1450-1516) fue un artista holandés cuyo cuadro más famoso es «El jardín de las delicias». El cuadro ilustra paneles exteriores e interiores . El panel exterior representa la creación del mundo, mientras que el interior consta de tres paneles que representan, respectivamente, El Jardín del Edén (panel izquierdo), El Mundo Humano (panel central) y El Juicio Final o Infierno (panel derecho). Un pequeño segmento del panel derecho ilustra un par de orejas en las que se ve una estructura que se asemeja a un PAuS (Figura 1) .

Figura1:El Bosco, El jardín de las delicias terrenales, una sección ampliada del panel derecho de la sección interior (El infierno). La oreja grotesca tiene una estructura que recuerda a un PauS (flecha).
PAuS, seno preauricular
Primeras descripciones médicas
El PAuS fue descrito por primera vez en 1864 por C. F. Heusinger al describir los hallazgos en un paciente característico del síndrome braquio-oto-renal (BOR) . Detalló su hallazgo y también se refirió a varios fenómenos ya descritos que parecían desvinculados entre sí, como el trabajo preliminar de Dzondi, que describió y definió las fístulas traqueales congénitas . Es importante señalar que la entidad de la aurícula descrita por Heusinger, aunque morfológicamente idéntica, es su entidad separada de las fístulas cervicales y otras fístulas braquiales descritas antes de sus observaciones.
Siguiendo la descripción de Heusinger, Rudolph Virchow (1821-1902) también describió la estructura en 1864, aunque limitándose a afirmar en su artículo «Yo también conozco un paciente así» . Sin embargo, Virchow fue el primero en postular que el PAuS es el resultado de un defecto en la fusión embriológica de los arcos faríngeos, afirmación que fue ampliamente contradicha en su momento (Figura 2).
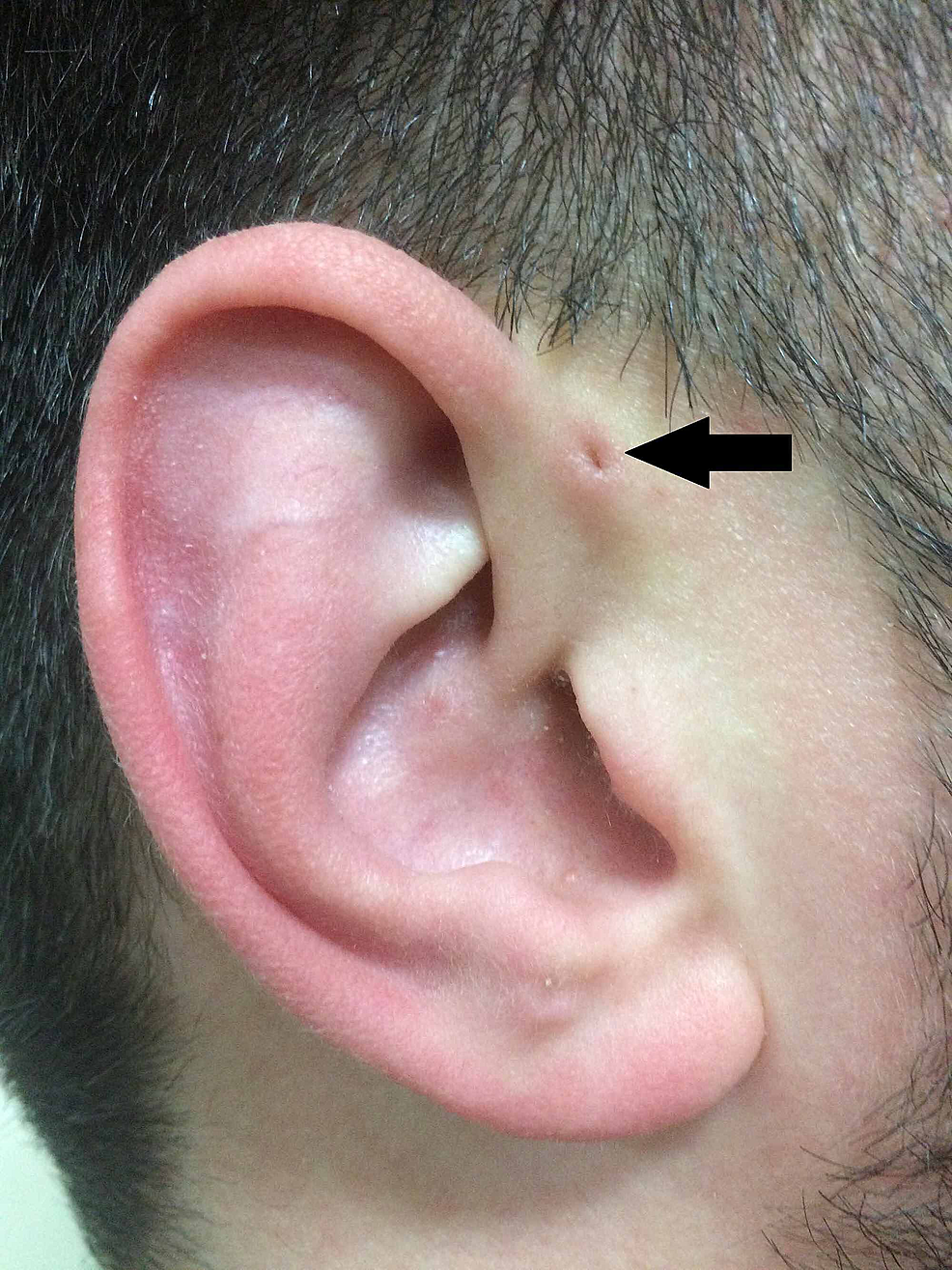
Figura2:Representación clásica del PAuS (flecha).
PAuS, seno preauricular
Sir James Paget también publicó sobre el tema en 1878, describiendo pacientes con este tipo de fístulas presentes no sólo en las orejas sino también en el cuello . Basándose en la descripción de sus pacientes, acuñó el término fístula oto-branquial (Figura 3). Sir James Paget adaptó ampliamente los puntos de vista de la teoría de Virchow sobre el origen de la PAuS.
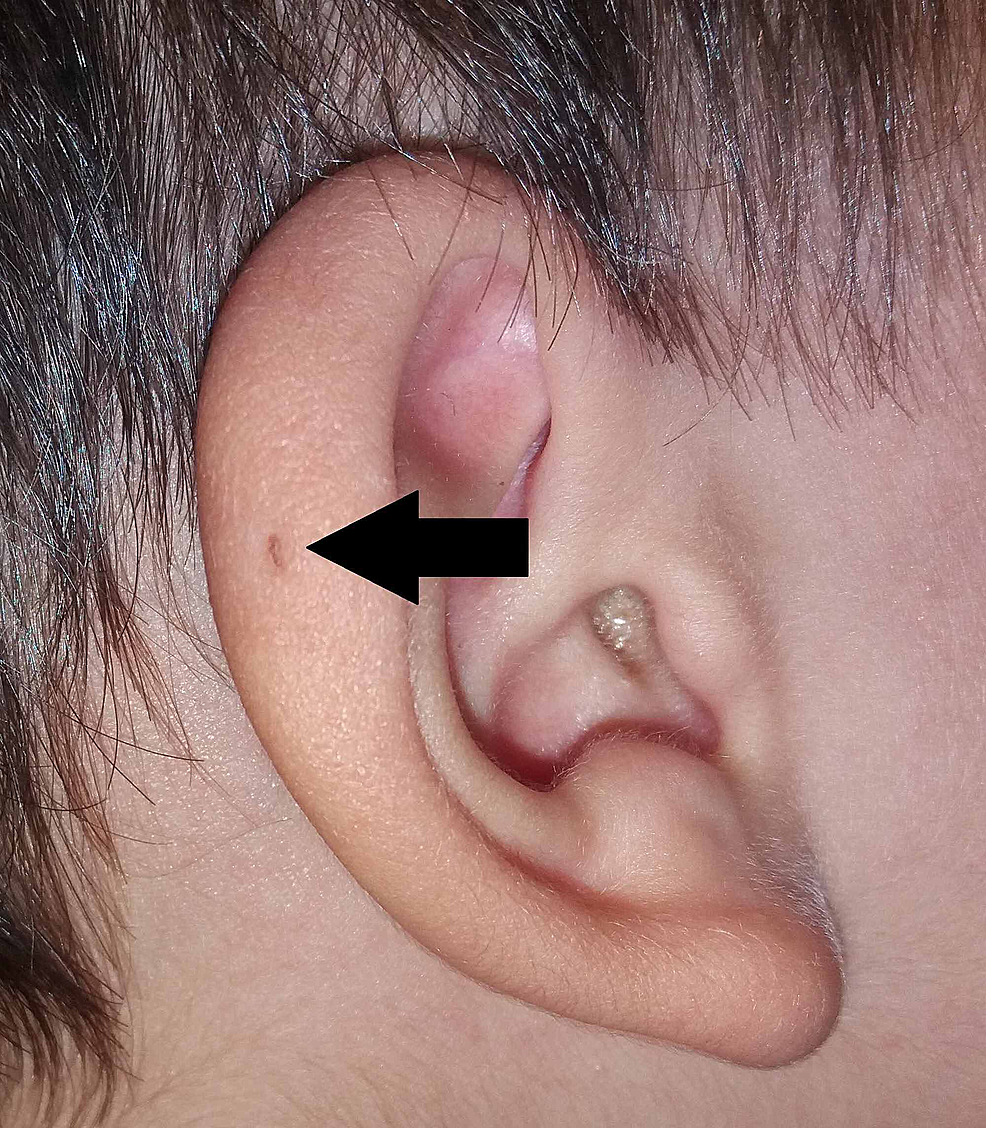
Figura3:Variante de PAuS en el hélix auricular (flecha).
PAuS, seno preauricular
Durante la primera parte del siglo XX, la PAuS se conoció como «agujeros de aretes naturales» y «fístula auris congénita», publicando varios autores sobre el tema, incluyendo su patrón de herencia . El fenómeno y su herencia transgeneracional permitieron un extenso conjunto de estudios y teorías sobre su causa, convergiendo ampliamente en las afirmaciones originales de Virchow.
Una de las contribuciones más importantes de ese período fue en 1955 por Fourman y Fourman, quienes investigaron la herencia de la PAuS, construyendo un árbol genealógico, centrándose en la herencia de la estructura . Llegaron a la conclusión de que la mitad de los hermanos de la familia afectada tienen la estructura y, por lo tanto, postularon que el seno se hereda como un rasgo dominante, pero con «penetrancia incompleta» debido a que se salta en las generaciones o en los individuos.
A pesar de ese interés inicial, sin embargo, se publicaron pocos o ningún artículo después de finales de la década de 1940, hasta un pequeño resurgimiento en los últimos años, donde la estructura reaparece bajo un nuevo nombre – PAuS. Desde entonces, rara vez se ha investigado la estructura, y los mecanismos de su herencia transgeneracional permanecen ampliamente olvidados en la literatura de los años 30 y 40.
Síndromes asociados al PAuS
Síndrome de Melnick-Fraser
El síndrome fue descrito por primera vez en 1864 por Heusinger . Sin embargo, en 1975, Melnick et al. describieron una serie de pacientes que lo padecían, y en 1980 Fraser et al. describieron otra serie de casos con las características del síndrome en una escuela especializada para sordos . La condición se considera relativamente rara, con 250 nuevos casos de Melnick-Fraser diagnosticados en Japón en 2014 . Primero se creyó que era una variante del síndrome BOR, pero más tarde Melnick amplió el síndrome a un grupo de afecciones casi idénticas que dependen de la presencia o ausencia de diferentes rasgos distintivos . El término síndrome de Melnick-Fraser se acuñó más tarde para unificar las características de los síndromes separados y para separarlo completamente del síndrome de Fraser, malformaciones urogenitales aisladas, y del síndrome de Frasier-Lynch, poliposis colorrectal familiar, con el que el BOR se había confundido muy a menudo como término en ese momento. Por lo tanto, lamentablemente la contribución de Heusinger ha sido ampliamente olvidada.
Síndrome de Beckwith-Wiedemann
La afección fue descrita originalmente en 1963 por el patólogo estadounidense John Beckwith como una combinación de exomphalos, macroglossia y gigantismo, por lo que se denominó síndrome de exomphalos-macroglossia-gigantism (EMG) (Poster: Beckwith, JB: Citomegalia extrema de la corteza fetal suprarrenal, hiperplasia de los riñones y del páncreas e hiperplasia de las células de Leydig : ¿Otro síndrome? Annual Meeting of Western Society for Pediatric Research, Los Ángeles, CA, 11 de noviembre de 1963). Sin embargo, en 1964, independientemente de John Beckwith, el pediatra alemán Hans-Rudolph Wiedemann también describió a varios pacientes con las mismas características de la enfermedad, pero que también incluían síntomas como la hiperplasia suprarrenal. Con el tiempo, el síndrome recibió el nombre de síndrome de Beckwith-Wiedemann, y los criterios de diagnóstico se ampliaron para incluir la PAuS, los riñones hiperplásicos, la microcefalia, la hipoglucemia neonatal y el hepatoblastoma que se desarrolla más adelante. En estos pacientes se han establecido mutaciones en 11p15 que afectan a genes como el factor de crecimiento similar a la insulina 2 (IGF-2), el inhibidor de la quinasa dependiente de ciclina 1C (CDKN1C), el H19 y el transcrito superpuesto 1 del canal de potasio de la subfamilia Q (KCNQ1OT1). El mecanismo de herencia aún no está definido, ya que en varios pacientes con esta afección no se ha podido determinar la herencia transgeneracional exacta, por lo que se considera un defecto esporádico recesivo. Es especialmente importante distinguir el síndrome de Beckwith-Wiedemann y sus características en los pacientes concebidos mediante fecundación in vitro, ya que la incidencia en esa población es mucho mayor. El síndrome de Beckwith-Wiedemann se considera una enfermedad rara con una incidencia de 1 por cada 13.700 niños nacidos, lo que supone un total de unos 300 niños nacidos con Beckwith-Wiedemann en los Estados Unidos de América anualmente.
Síndrome de Lachiewicz-Sibley
Descrito por primera vez en 1985 por Lachiewicz et al., es uno de los síndromes más raros jamás descritos . El síndrome es muy similar al de Melnick-Fraser, aunque sólo se encontró PAuS y riñones hipoplásicos con proteinuria de inicio temprano en los descendientes de inmigrantes británicos e irlandeses que se asentaron en Ohio en el siglo XIX y posteriormente en Nebraska. En el momento del estudio original, de 130 parientes vivos, 12 miembros tenían PAuS y riñones hipoplásicos, 10 sólo tenían riñones hipoplásicos y 3 sólo tenían PAuS . Aunque el locus exacto de la mutación sigue siendo desconocido, la afección se hereda de forma autosómica dominante.
Siglo XXI
Hoy en día, aunque todavía está relativamente poco investigado, el PAuS es una entidad clínica común con estrategias clínicas bien definidas para su tratamiento. Algunos estudios han identificado una asociación genética entre el PAuS y el locus 8q11.1-13.1; sin embargo, los resultados no se han reproducido ampliamente . Aparte de este estudio individual, ningún otro estudio se ha centrado en la razón genética del SSPA ni lo ha asociado con los síndromes genéticos mencionados, y los estudios genealógicos son escasos y poco frecuentes.
El curso clínico ha sido bien descrito, y las estrategias de tratamiento, que incluyen predominantemente la escisión quirúrgica, han sido bien establecidas en la otorrinolaringología y la cirugía de cabeza y cuello modernas. Hoy en día, los pacientes con PAuS, no asociados a un síndrome genético, se someten a un proceso de intervención seguro con pocas complicaciones descritas, mientras que los pacientes con síndromes genéticos se someten a las mismas modalidades curativas, y la escisión del PauS no afecta a su curso clínico general .