- Rappresentazione artistica
- Figura1:Hieronymous Bosch, Garden of Earthly Delights, una sezione ingrandita del pannello destro della sezione interna (Inferno). L’orecchio grottesco ha una struttura che ricorda un PauS (freccia).
- Prime descrizioni mediche
- Figura2:Rappresentazione classica della PAuS (freccia).
- Figura3:Variante della PAuS sull’elice auricolare (freccia).
- Sindromi associate a PAuS
- Sindrome di Melnick-Fraser
- Sindrome di Beckwith-Wiedemann
- Sindrome di Lachiewicz-Sibley
- Ventunesimo secolo
Rappresentazione artistica
Hieronymus Bosh (c. 1450-1516) era un artista olandese il cui dipinto più famoso è “The Garden of Earthly Delights”. Il dipinto illustra pannelli esterni e interni. Il pannello esterno ricorda la creazione del mondo, mentre l’interno è composto da tre pannelli che rappresentano, rispettivamente, Il giardino dell’Eden (pannello di sinistra), Il mondo umano (pannello centrale), e Il giorno del giudizio o inferno (pannello di destra). Un piccolo segmento del pannello di destra illustra un paio di orecchie su cui si vede una struttura simile a un PAuS (Figura 1).

Figura1:Hieronymous Bosch, Garden of Earthly Delights, una sezione ingrandita del pannello destro della sezione interna (Inferno). L’orecchio grottesco ha una struttura che ricorda un PauS (freccia).
PAuS, seno preauricolare
Prime descrizioni mediche
Il PAuS fu descritto per la prima volta nel 1864 da C. F. Heusinger descrivendo i risultati in un paziente caratteristico della sindrome brachio-oto-renale (BOR) . Egli descrisse dettagliatamente la sua scoperta e fece anche riferimento a diversi fenomeni già descritti che sembravano distaccati l’uno dall’altro, come il lavoro preliminare di Dzondi, che ha descritto e definito le fistole tracheali congenite. È importante notare che l’entità auricolare descritta da Heusinger, anche se morfologicamente identica, è la sua entità separata dalle fistole cervicali e altre fistole brachiali descritte prima delle sue osservazioni .
Seguendo la descrizione di Heusinger, Rudolph Virchow (1821-1902) ha anche descritto la struttura nel 1864, anche se semplicemente affermando nel suo articolo “So anche un paziente come questo” . Virchow, tuttavia, fu il primo a postulare che la PAuS è il risultato di un difetto nella fusione embriologica delle arcate faringee, un’affermazione che fu ampiamente contraddetta all’epoca (Figura 2).
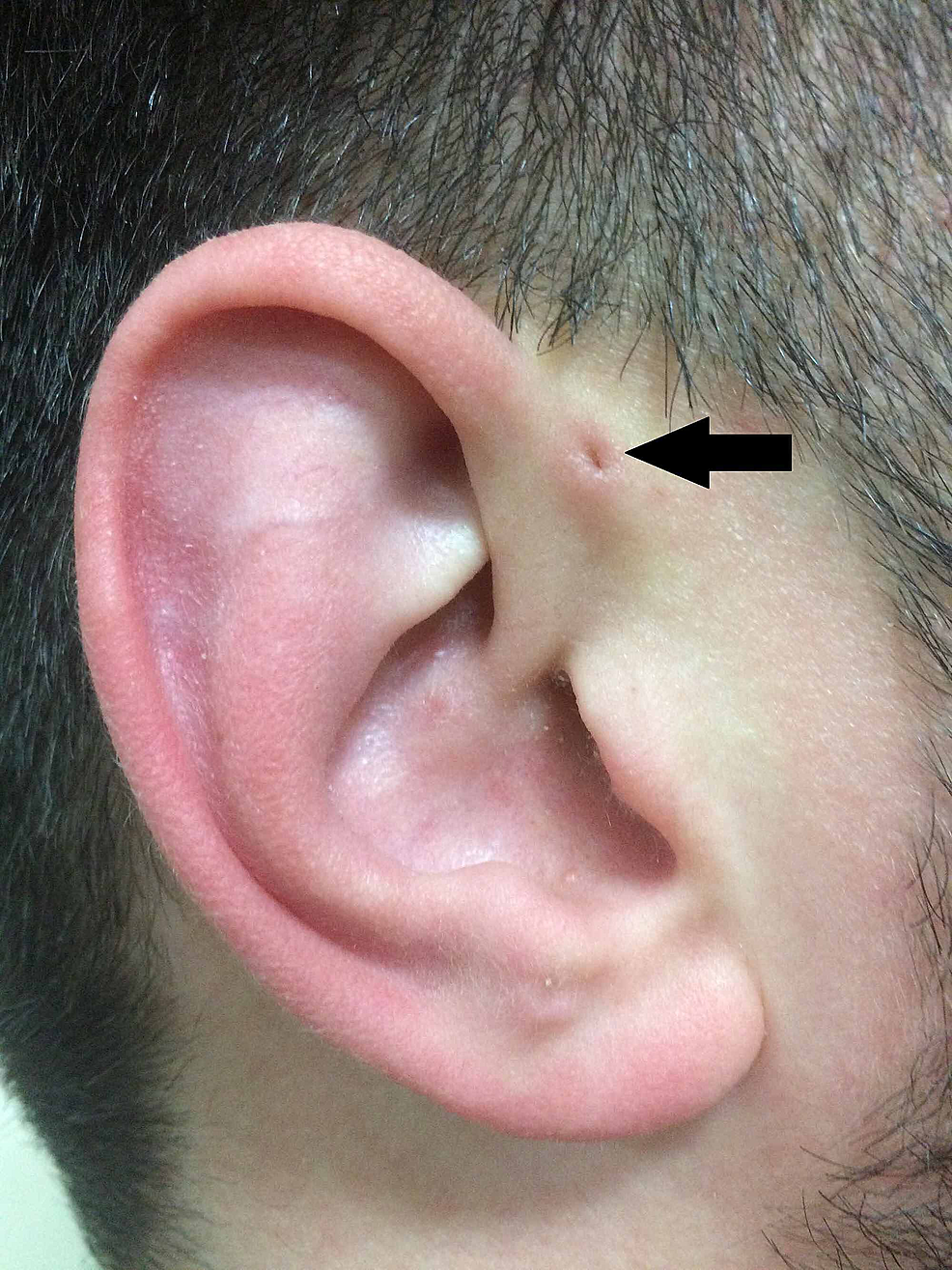
Figura2:Rappresentazione classica della PAuS (freccia).
PAuS, seno preauricolare
Anche Sir James Paget pubblicò sull’argomento nel 1878, descrivendo pazienti con tali fistole presenti non solo sulle orecchie ma anche sul collo. Sulla base della descrizione dei suoi pazienti, egli coniò il termine fistola oto-branchiale (Figura 3). Sir James Paget adattò ampiamente i punti di vista della teoria di Virchow sull’origine della PAuS.
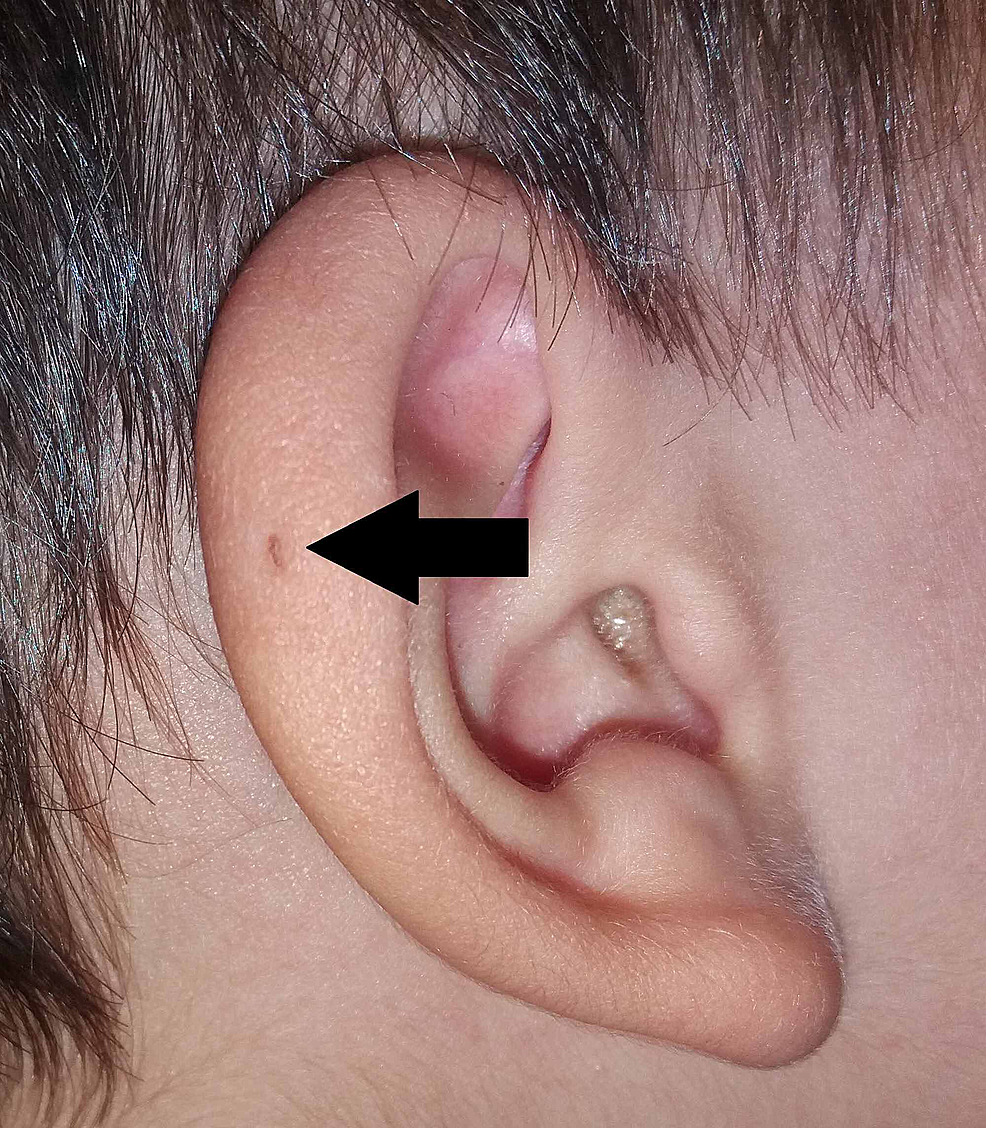
Figura3:Variante della PAuS sull’elice auricolare (freccia).
PAuS, seno preauricolare
Durante la prima parte del XX secolo, il PAuS divenne noto come “buchi naturali dell’orecchio” e “fistola auris congenita”, con diversi autori che pubblicarono sull’argomento, incluso il suo modello di eredità. Il fenomeno e la sua eredità transgenerazionale hanno permesso una vasta serie di studi e teorie sulla sua causa, ampiamente convergenti sulle dichiarazioni originali di Virchow.
Uno dei contributi più importanti di quel periodo è stato nel 1955 da Fourman e Fourman, che hanno studiato l’eredità della PAuS, costruendo un albero genealogico, concentrandosi sull’eredità della struttura. Hanno concluso che la metà dei fratelli della famiglia colpita ha la struttura e quindi postulato che il seno è ereditato come un tratto dominante, ma con “penetranza incompleta” a causa di saltare in generazioni o individui.
Nonostante questo interesse iniziale, tuttavia, poco o nessun articolo è stato pubblicato dopo la fine degli anni ’40, fino a una piccola ripresa negli anni successivi, dove la struttura riappare con un nuovo nome – PAuS. Da allora, la ricerca sulla struttura è stata effettuata raramente, e i meccanismi della sua eredità transgenerazionale rimangono ampiamente dimenticati nella letteratura degli anni ’30 e ’40.
Sindromi associate a PAuS
Sindrome di Melnick-Fraser
La sindrome fu descritta per la prima volta nel 1864 da Heusinger . Tuttavia, nel 1975, Melnick et al. hanno descritto una serie di pazienti con essa, e nel 1980 Fraser et al. hanno descritto un’altra serie di casi con le caratteristiche della sindrome in una scuola specializzata per sordi. La condizione è considerata relativamente rara, con 250 nuovi casi di Melnick-Fraser diagnosticati in Giappone nel 2014. Inizialmente ritenuto una variante della sindrome di BOR, in seguito Melnick ha ampliato la sindrome in un gruppo di condizioni quasi identiche dipendenti dalla presenza o assenza di diversi segni distintivi. Il termine sindrome Melnick-Fraser fu poi coniato per unificare le caratteristiche delle sindromi separate e per separarla completamente dalla sindrome di Fraser, malformazioni urogenitali isolate, e dalla sindrome di Frasier-Lynch, poliposi colorettale familiare, con la quale BOR era stata spesso confusa come termine in quel momento. Così, purtroppo il contributo di Heusinger è stato ampiamente dimenticato.
Sindrome di Beckwith-Wiedemann
La condizione è stata originariamente descritta nel 1963 dal patologo americano John Beckwith come una combinazione di esomphalos, macroglossia e gigantismo, quindi indicato come exomphalos-macroglossia-gigantism (EMG) syndrome (Poster: Beckwith, JB: Citomegalia estrema della corteccia surrenale fetale, iperplasia dei reni e del pancreas, e iperplasia delle cellule di Leydig: Un’altra sindrome? Annual Meeting of Western Society for Pediatric Research, Los Angeles, CA, November 11, 1963). Nel 1964, tuttavia, indipendentemente da John Beckwith, il pediatra tedesco Hans-Rudolph Wiedemann ha anche descritto diversi pazienti con le stesse caratteristiche della malattia, ma anche sintomi come l’iperplasia surrenale. Nel corso del tempo, la sindrome è stata soprannominata sindrome di Beckwith-Wiedemann, e i criteri diagnostici sono stati ampliati per includere PAuS, reni iperplastici, microcefalia, ipoglicemia neonatale, ed epatoblastoma che si sviluppa più tardi nella vita. Mutazioni in 11p15 che coinvolgono geni come il fattore di crescita insulino-simile 2 (IGF-2), inibitore della chinasi ciclina-dipendente 1C (CDKN1C), H19, e potassio voltaggio-gated channel subfamily Q member 1 overlapping transcript 1 (KCNQ1OT1) sono stati stabiliti in tali pazienti. Il meccanismo di eredità è ancora indefinito in quanto in diversi pazienti con questa condizione, l’esatta eredità transgenerazionale non potrebbe essere determinata, ritenendola un difetto sporadico recessivo. La sindrome di Beckwith-Wiedemann e le sue caratteristiche sono particolarmente importanti da distinguere nei pazienti concepiti con la fecondazione in vitro in quanto l’incidenza in quella popolazione è molto più alta. Beckwith-Wiedemann è considerata una condizione rara con un’incidenza di 1 ogni 13.700 bambini nati, per un totale di circa 300 bambini nati con Beckwith-Wiedemann negli Stati Uniti d’America ogni anno.
Sindrome di Lachiewicz-Sibley
Descritta per la prima volta nel 1985 da Lachiewicz e altri, questa è una delle sindromi più rare mai descritte. La sindrome è molto simile alla Melnick-Fraser, anche se solo la PAuS e i reni ipoplasici con proteinuria precoce sono stati trovati nei discendenti degli immigrati britannici e irlandesi che si stabilirono in Ohio nel 1800 e poi in Nebraska. All’epoca dello studio originale, su 130 parenti viventi, 12 membri avevano PAuS e reni ipoplasici, 10 avevano solo reni ipoplasici, e 3 avevano solo PAuS . Anche se il locus esatto della mutazione rimane sconosciuto, la condizione è ereditata in modo autosomico dominante.
Ventunesimo secolo
Oggi, sebbene ancora relativamente poco studiato, la PAuS è un’entità clinica comune con strategie cliniche ben definite per il trattamento. Alcuni studi hanno identificato un’associazione genetica tra la PAuS e il locus 8q11.1-13.1; tuttavia, i risultati non sono stati riprodotti ampiamente. Oltre a questo studio individuale, nessun altro studio si è concentrato sulla ragione genetica della PAuS né l’ha associata alle sindromi genetiche sopra menzionate, con studi genealogici che sono pochi e lontani tra loro.
Il decorso clinico è stato ben descritto, e le strategie di trattamento che includono prevalentemente l’escissione chirurgica sono state ben stabilite nella moderna otorinolaringoiatria e chirurgia della testa e del collo. Al giorno d’oggi, i pazienti con PAuS, non associati a una sindrome genetica, sono sottoposti a un processo di intervento sicuro con poche complicazioni descritte, mentre i pazienti con sindromi genetiche sono sottoposti alle stesse modalità curative, e l’escissione della PauS non influenza il loro corso clinico complessivo .
.