- Représentation artistique
- Figure1:Hieronymous Bosch, Le Jardin des délices terrestres, une section agrandie du panneau de droite de la section intérieure (Enfer). L’oreille grotesque a une structure rappelant celle d’un PauS (flèche).
- Premières descriptions médicales
- Figure2:Représentation classique du PAuS (flèche).
- Figure3:Variante du PAuS sur l’hélice auriculaire (flèche).
- Syndromes associés au PAuS
- Syndrome de Melnick-Fraser
- Syndrome de Beckwith-Wiedemann
- Syndrome de Lachiewicz-Sibley
- Vingt-et-unième siècle
Représentation artistique
Hieronymus Bosh (vers 1450-1516) était un artiste néerlandais dont le tableau le plus célèbre est « Le jardin des délices terrestres ». La peinture illustre des panneaux extérieurs et intérieurs . Le panneau extérieur ressemble à la création du monde, tandis que l’intérieur se compose de trois panneaux qui représentent, respectivement, le jardin d’Eden (panneau de gauche), le monde humain (panneau du milieu) et le jugement dernier ou l’enfer (panneau de droite). Un petit segment du panneau de droite illustre une paire d’oreilles sur lesquelles on peut voir une structure ressemblant à un PAuS (Figure 1) .

Figure1:Hieronymous Bosch, Le Jardin des délices terrestres, une section agrandie du panneau de droite de la section intérieure (Enfer). L’oreille grotesque a une structure rappelant celle d’un PauS (flèche).
PAuS, sinus préauriculaire
Premières descriptions médicales
Le PAuS a été décrit pour la première fois en 1864 par C. F. Heusinger lorsqu’il a décrit les résultats chez un patient caractéristique du syndrome brachio-oto-rénal (BOR) . Il a détaillé sa découverte et a également fait référence à plusieurs phénomènes déjà décrits qui semblaient détachés les uns des autres, tels que les travaux préliminaires de Dzondi, qui a décrit et défini les fistules trachéales congénitales . Il est important de noter que l’entité auriculaire décrite par Heusinger, bien que morphologiquement identique, est son entité distincte des fistules cervicales et autres fistules brachiales décrites avant ses observations .
Suivant la description de Heusinger, Rudolph Virchow (1821-1902) a également décrit la structure en 1864, bien que déclarant simplement dans son article « Je connais aussi un patient comme ça » . Virchow a cependant été le premier à postuler que le PAuS résulte d’un défaut de fusion embryologique des arcs pharyngés, une affirmation largement contredite à l’époque (figure 2).
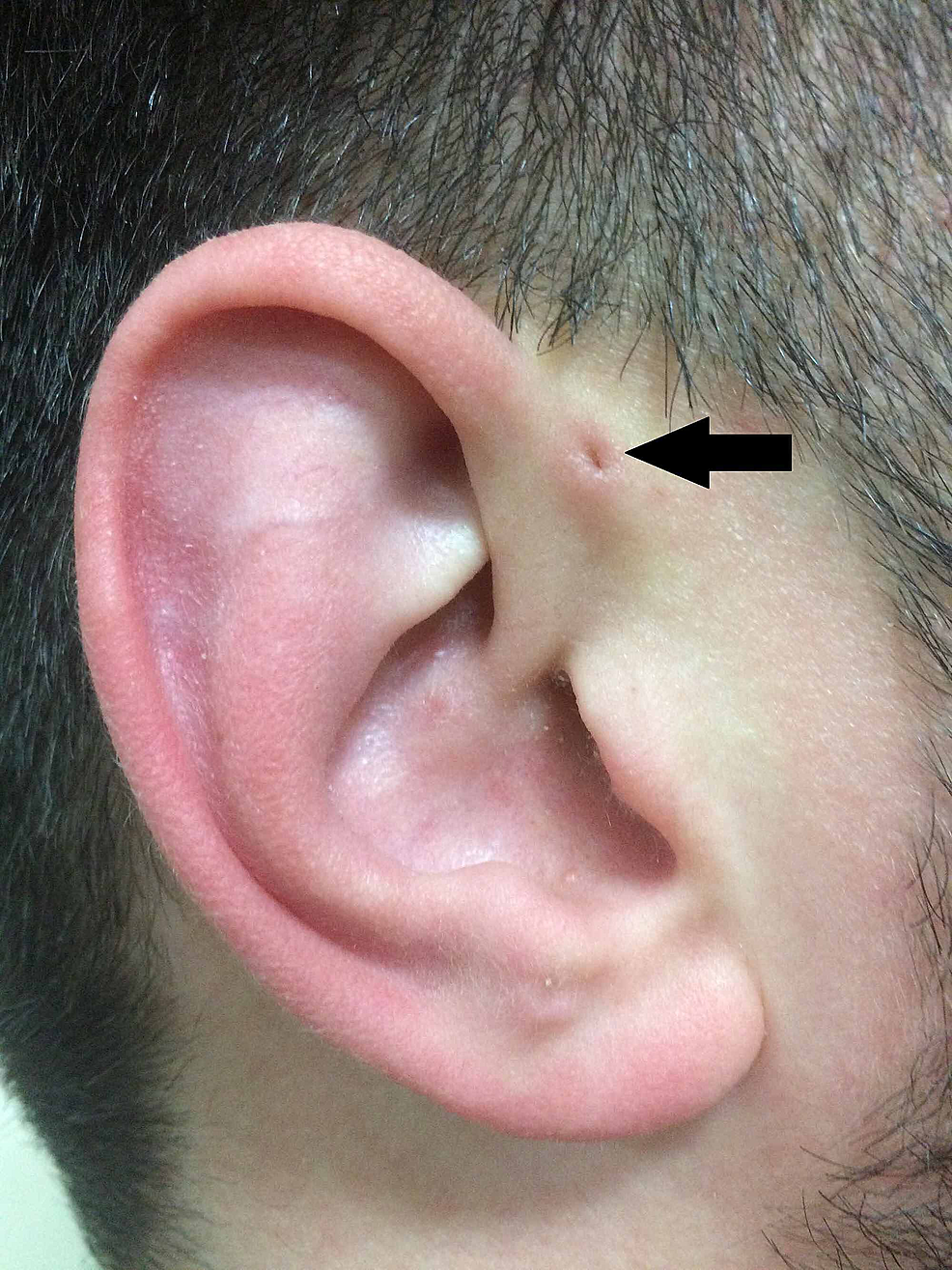
Figure2:Représentation classique du PAuS (flèche).
PAuS, sinus préauriculaire
Sir James Paget a également publié sur le sujet en 1878, décrivant des patients avec de telles fistules présentes non seulement sur leurs oreilles mais aussi sur leur cou . Sur la base de la description de ses patients, il a inventé le terme de fistule oto-branchiale (figure 3). Sir James Paget a largement adapté les points de vue de la théorie de Virchow sur l’origine du PAuS.
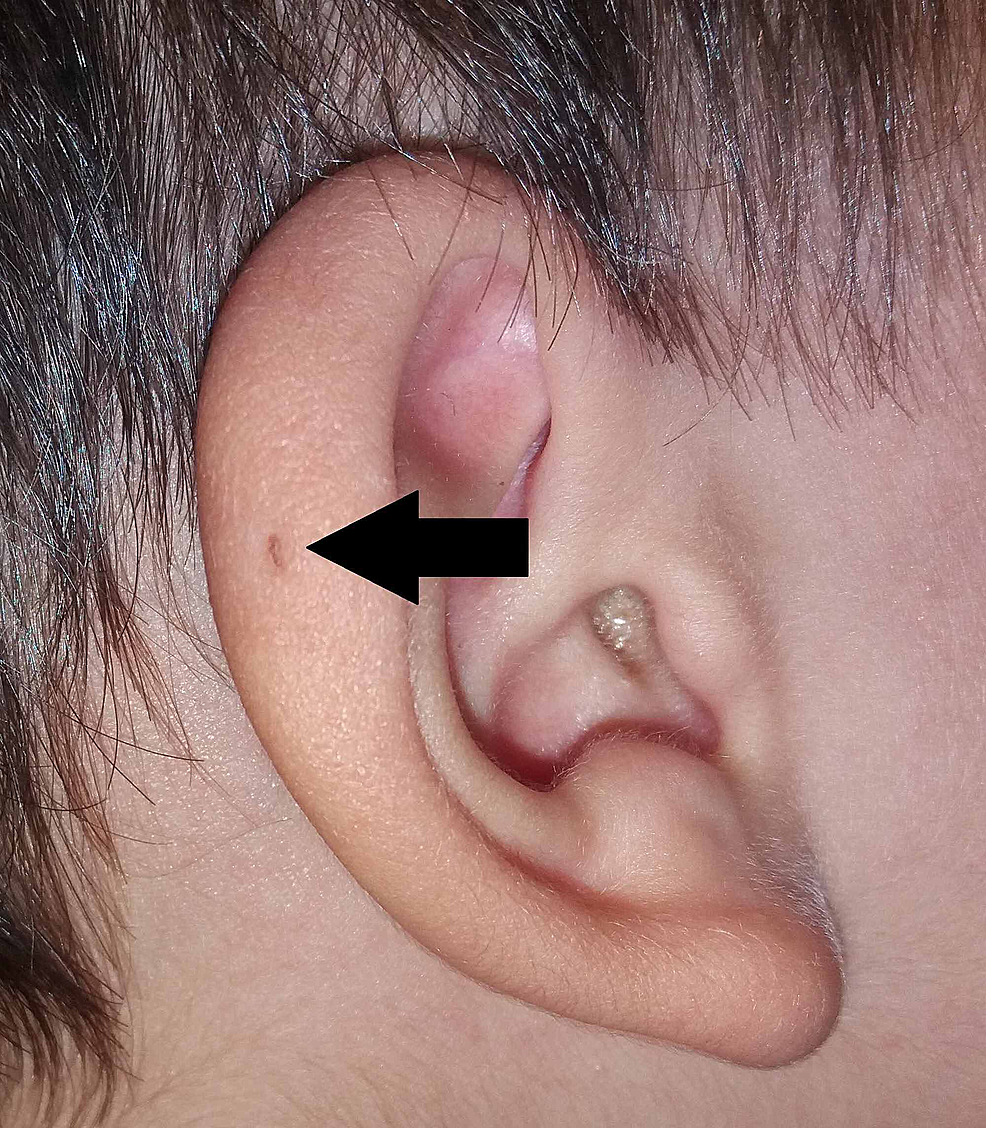
Figure3:Variante du PAuS sur l’hélice auriculaire (flèche).
PAuS, sinus préauriculaire
Durant la première partie du 20ème siècle, le PAuS est devenu connu sous le nom de « trous de boucle d’oreille naturels » et de « fistule auris congenita », avec plusieurs auteurs publiant sur le sujet, y compris son modèle d’héritage . Le phénomène et sa transmission transgénérationnelle ont donné lieu à un grand nombre d’études et de théories sur sa cause, qui convergeaient largement vers les déclarations originales de Virchow.
L’une des contributions les plus importantes de cette période a été réalisée en 1955 par Fourman et Fourman, qui ont étudié la transmission du PAuS, en construisant un arbre généalogique, en se concentrant sur la transmission de la structure . Ils ont conclu que la moitié des frères et sœurs de la famille affectée ont la structure et ont donc postulé que le sinus est hérité comme un trait dominant, mais avec une « pénétrance incomplète » en raison de son saut dans les générations ou les individus .
Malgré cet intérêt initial, cependant, peu ou pas d’articles ont été publiés après la fin des années 1940, jusqu’à une petite résurgence dans les années ultérieures, où la structure réapparaît sous un nouveau nom – PAuS. Depuis lors, les recherches sur cette structure ont rarement été menées, et les mécanismes de son héritage transgénérationnel restent largement oubliés dans la littérature des années 1930 et 1940.
Syndromes associés au PAuS
Syndrome de Melnick-Fraser
Le syndrome a été décrit pour la première fois en 1864 par Heusinger . Cependant, en 1975, Melnick et al. ont décrit une série de patients qui en étaient atteints, et en 1980, Fraser et al. ont décrit une autre série de cas présentant les caractéristiques du syndrome dans une école spécialisée pour les sourds . Cette affection est considérée comme relativement rare, 250 nouveaux cas de Melnick-Fraser ayant été diagnostiqués au Japon en 2014 . D’abord considéré comme une variante du syndrome BOR, Melnick a ensuite élargi le syndrome à un groupe d’affections presque identiques dépendant de la présence ou de l’absence de différentes caractéristiques. Le terme de syndrome de Melnick-Fraser a ensuite été inventé pour unifier les caractéristiques des syndromes distincts et pour le séparer complètement du syndrome de Fraser, malformations urogénitales isolées, et du syndrome de Frasier-Lynch, polypose colorectale familiale, avec lesquels le syndrome de BOR avait été assez souvent confondu à l’époque. Ainsi, malheureusement, la contribution de Heusinger a été largement oubliée.
Syndrome de Beckwith-Wiedemann
L’affection a été décrite à l’origine en 1963 par le pathologiste américain John Beckwith comme une combinaison d’exomphalie, de macroglossie et de gigantisme, donc appelée syndrome d’exomphalie-macroglossie-gigantisme (EMG) (Poster : Beckwith, JB : Cytomégalie extrême du cortex fœtal surrénalien, hyperplasie des reins et du pancréas, et hyperplasie des cellules de Leydig : Un autre syndrome ? Réunion annuelle de la Western Society for Pediatric Research, Los Angeles, CA, 11 novembre 1963). En 1964, cependant, indépendamment de John Beckwith, le pédiatre allemand Hans-Rudolph Wiedemann a également décrit plusieurs patients présentant les mêmes caractéristiques de la maladie mais incluant également des symptômes tels que l’hyperplasie des surrénales . Au fil du temps, le syndrome a été baptisé syndrome de Beckwith-Wiedemann, et les critères de diagnostic ont été élargis pour inclure le PAuS, l’hyperplasie rénale, la microcéphalie, l’hypoglycémie néonatale et l’hépatoblastome se développant plus tard dans la vie . Des mutations dans le gène 11p15 impliquant des gènes tels que le facteur de croissance analogue à l’insuline 2 (IGF-2), l’inhibiteur de kinase dépendant de la cycline 1C (CDKN1C), H19 et le transcrit superposé 1 du canal potassique voltage-gated membre de la sous-famille Q (KCNQ1OT1) ont été établies chez ces patients. Le mécanisme d’hérédité n’est toujours pas défini, car chez plusieurs patients atteints de cette maladie, la transmission transgénérationnelle exacte n’a pu être déterminée, ce qui en fait un défaut récessif sporadique. Il est particulièrement important de distinguer le syndrome de Beckwith-Wiedemann et ses caractéristiques chez les patients conçus par fécondation in vitro, car l’incidence dans cette population est beaucoup plus élevée. Beckwith-Wiedemann est considéré comme une condition rare avec une incidence de 1 pour 13 700 enfants nés, ce qui représente un total d’environ 300 enfants nés avec Beckwith-Wiedemann aux États-Unis d’Amérique chaque année .
Syndrome de Lachiewicz-Sibley
Décrit pour la première fois en 1985 par Lachiewicz et al, c’est l’un des syndromes les plus rares jamais décrits . Le syndrome est très similaire à celui de Melnick-Fraser, bien que seuls le PAuS et les reins hypoplastiques avec une protéinurie à début précoce aient été trouvés chez les descendants d’immigrants britanniques et irlandais s’installant dans l’Ohio dans les années 1800 et plus tard dans le Nebraska. Au moment de l’étude originale, sur 130 parents vivants, 12 membres avaient le PAuS et des reins hypoplasiques, 10 avaient seulement des reins hypoplasiques et 3 avaient seulement le PAuS . Bien que le locus exact de la mutation reste inconnu, l’affection est héritée sur un mode autosomique dominant.
Vingt-et-unième siècle
Aujourd’hui, bien qu’encore relativement peu étudié, le PAuS est une entité clinique courante avec des stratégies cliniques de traitement bien définies. Certaines études ont identifié une association génétique entre le PAuS et le locus 8q11.1-13.1 ; cependant, les résultats n’ont pas été largement reproduits . En dehors de cette étude individuelle, aucune autre étude ne s’est concentrée sur la raison génétique du PAuS ni ne l’a associé aux syndromes génétiques susmentionnés, les études généalogiques étant peu nombreuses .
L’évolution clinique a été bien décrite, et les stratégies de traitement, y compris l’excision chirurgicale prédominante, ont été bien établies dans l’otorhinolaryngologie et la chirurgie de la tête et du cou modernes . Aujourd’hui, les patients atteints de PAuS, non associés à un syndrome génétique, subissent un processus d’intervention sûr avec peu de complications décrites, tandis que les patients atteints de syndromes génétiques subissent les mêmes modalités curatives, et l’excision du PauS n’affecte pas leur évolution clinique globale .
.