- Opracowanie artystyczne
- Rysunek1:Hieronymous Bosch, Ogród rozkoszy ziemskich, powiększony fragment prawego panelu części wewnętrznej (Piekło). Groteskowe ucho ma strukturę przypominającą PauS (strzałka).
- Pierwsze opisy medyczne
- Rycina 2:Klasyczne przedstawienie PAuS (strzałka).
- Rycina 3:Wariant PAuS na spirali małżowiny usznej (strzałka).
- Zespoły związane z PAuS
- Syndrom Melnicka-Frasera
- Syndrom Beckwitha-Wiedemanna
- Zespół Lachiewicza-Sibleya
- Dwudziesty pierwszy wiek
Opracowanie artystyczne
Hieronymus Bosh (ok. 1450-1516) był holenderskim artystą, którego najsłynniejszym obrazem jest „Ogród rozkoszy ziemskich”. Obraz ten ilustruje panele zewnętrzne i wewnętrzne. Panel zewnętrzny przypomina stworzenie świata, natomiast wnętrze składa się z trzech paneli, które przedstawiają odpowiednio: rajski ogród (panel lewy), świat ludzi (panel środkowy) oraz dzień sądu lub piekło (panel prawy). Niewielki fragment prawego panelu przedstawia parę uszu, na których widoczna jest struktura przypominająca PAuS (Rysunek 1) .

Rysunek1:Hieronymous Bosch, Ogród rozkoszy ziemskich, powiększony fragment prawego panelu części wewnętrznej (Piekło). Groteskowe ucho ma strukturę przypominającą PauS (strzałka).
PAuS, preauricular sinus
Pierwsze opisy medyczne
PauS został po raz pierwszy opisany w 1864 r. przez C. F. Heusingera podczas opisywania znalezisk u pacjenta charakterystycznych dla zespołu brachio-oto-renal (BOR) . Szczegółowo opisał on swoje odkrycie, a także odniósł się do kilku już opisanych zjawisk, które wydawały się od siebie oderwane, takich jak wstępna praca Dzondiego, który opisał i zdefiniował wrodzone przetoki tchawicze. Ważne jest, aby zauważyć, że jednostka auricula opisana przez Heusingera, choć morfologicznie identyczna, jest jej odrębną jednostką od przetok szyjnych i innych przetok ramiennych opisanych przed jego obserwacjami .
Po opisie Heusingera, Rudolph Virchow (1821-1902) również opisał tę strukturę w 1864 roku, choć po prostu stwierdzając w swoim artykule „Znam też takiego pacjenta” . Virchow był jednak pierwszym, który postulował, że PAuS jest wynikiem defektu w embriologicznym połączeniu łuków gardłowych, co w tamtym czasie było powszechnie negowane (Rycina 2).
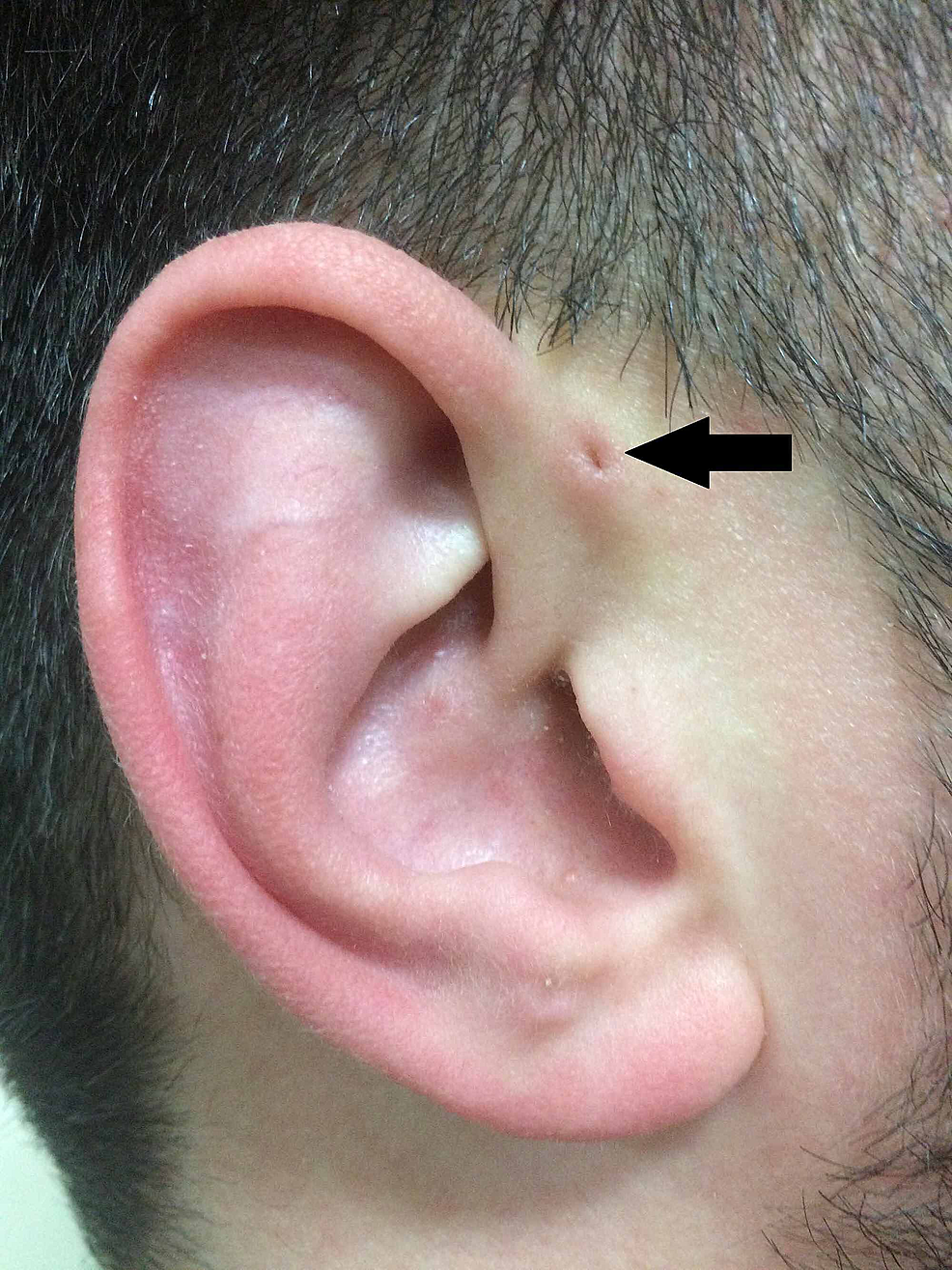
Rycina 2:Klasyczne przedstawienie PAuS (strzałka).
PAuS, preauricular sinus
Sir James Paget również opublikował na ten temat w 1878 roku, opisując pacjentów z takimi przetokami obecnymi nie tylko na uszach, ale również na szyi. Na podstawie opisu swoich pacjentów ukuł termin oto-branchial fistula (ryc. 3). Sir James Paget szeroko zaadaptował poglądy Virchowa dotyczące pochodzenia PAuS.
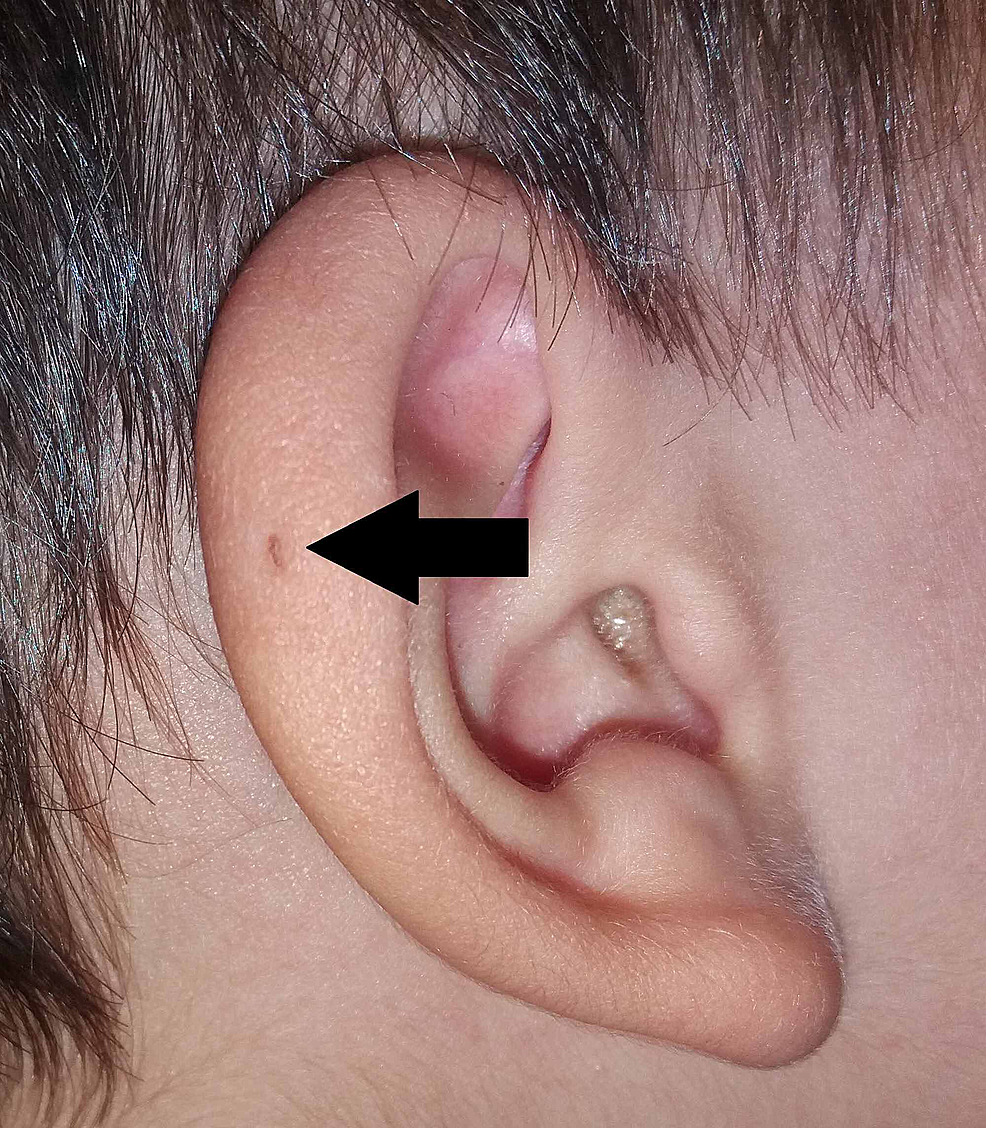
Rycina 3:Wariant PAuS na spirali małżowiny usznej (strzałka).
PAuS, preauricular sinus
W pierwszej połowie XX wieku PAuS stał się znany jako „naturalne otwory kolczykowate” i „fistula auris congenita”, a kilku autorów publikowało na ten temat, w tym na temat jego wzoru dziedziczenia. Zjawisko i jego transgeneracyjne dziedziczenie pozwoliło na obszerny zestaw badań i teorii na temat jego przyczyny, powszechnie zbieżne z oryginalnymi stwierdzeniami Virchowa .
Jeden z najważniejszych wkładów z tego okresu czasu był w 1955 roku przez Fourman i Fourman, którzy badali dziedziczenie PAuS, konstruując drzewo genealogiczne, koncentrując się na dziedziczeniu struktury . Doszli oni do wniosku, że połowa rodzeństwa rodziny dotkniętej chorobą ma tę strukturę i w ten sposób postulowali, że zatoka jest dziedziczona jako cecha dominująca, ale z „niepełną penetracją” z powodu jej pomijania w pokoleniach lub osobnikach .
Mimo tego początkowego zainteresowania, jednak niewiele do żadnych artykułów zostało opublikowanych po późnych latach 40-tych, aż do małego odrodzenia w późniejszych latach, gdzie struktura pojawia się ponownie pod nową nazwą – PAuS. Od tego czasu badania nad tą strukturą są rzadko prowadzone, a mechanizmy jej transgeneracyjnego dziedziczenia pozostają powszechnie zapomniane w literaturze z lat 30. i 40. ubiegłego wieku.
Zespoły związane z PAuS
Syndrom Melnicka-Frasera
Zespół ten został po raz pierwszy opisany w 1864 roku przez Heusingera . Jednak w 1975 roku Melnick i wsp. opisali serię pacjentów z tym zespołem, a w 1980 roku Fraser i wsp. opisali kolejną serię przypadków z cechami zespołu w specjalistycznej szkole dla głuchych. Warunek jest uważany za stosunkowo rzadki, a 250 nowych przypadków Melnick-Fraser zostało zdiagnozowanych w Japonii w 2014 roku . Najpierw uważany za wariant zespołu BOR, później Melnick rozszerzył zespół na grupę niemal identycznych warunków zależnych od obecności lub braku różnych cech charakterystycznych . Termin zespół Melnick-Fraser został później ukuty w celu ujednolicenia cech oddzielnych zespołów i całkowitego oddzielenia go od zespołu Frasera, izolowanych wad rozwojowych układu moczowo-płciowego, i zespołu Frasiera-Lyncha, rodzinnej polipowatości jelita grubego, z którymi BOR był dość często mylony jako termin w tym momencie. Dlatego też, niestety, wkład Heusingera został powszechnie zapomniany.
Syndrom Beckwitha-Wiedemanna
Stan ten został pierwotnie opisany w 1963 roku przez amerykańskiego patologa Johna Beckwitha jako połączenie egzomphalos, makroglosji i gigantyzmu, dlatego też określany jest jako zespół egzomphalos-macroglossia-gigantism (EMG) (Poster: Beckwith, JB: Extreme Cytomegaly of the adrenal Fetal Cortex, Hyperplasia of the Kidneys and Pancreas, and Leydig-Cell Hyperplasia : Another Syndrome? Annual Meeting of Western Society for Pediatric Research, Los Angeles, CA, 11 listopada 1963). W 1964 roku, jednak niezależnie od Johna Beckwitha, niemiecki pediatra Hans-Rudolph Wiedemann również opisał kilku pacjentów z tymi samymi cechami choroby, ale również zawierał objawy, takie jak hiperplazja nadnerczy . Z czasem zespół został nazwany zespołem Beckwitha-Wiedemanna, a kryteria diagnostyczne zostały rozszerzone o PAuS, hiperplastyczne nerki, mikrocefalię, hipoglikemię noworodkową i hepatoblastomę rozwijającą się w późniejszym okresie życia. Mutacje w 11p15 obejmujące takie geny jak insulinopodobny czynnik wzrostu 2 (IGF-2), inhibitor kinazy zależnej od cyklin 1C (CDKN1C), H19 oraz podrodzinę 1 nakładającego się transkryptu 1 (KCNQ1OT1) kanału bramkowanego napięciem potasowym (potassium voltage-gated channel subfamily Q member 1 overlapping transcript 1) zostały stwierdzone u tych pacjentów. Mechanizm dziedziczenia jest nadal nieokreślony, ponieważ u kilku pacjentów z tym schorzeniem nie udało się ustalić dokładnego dziedziczenia transgeneracyjnego, uznając je za sporadyczny defekt recesywny. Zespół Beckwitha-Wiedemanna i jego cechy charakterystyczne są szczególnie ważne do wyróżnienia u pacjentów poczętych w wyniku zapłodnienia in vitro, ponieważ częstość występowania w tej populacji jest znacznie wyższa. Zespół Beckwitha-Wiedemanna jest uważany za rzadkie schorzenie, którego częstość występowania wynosi 1 na 13 700 urodzonych dzieci, co odpowiada około 300 dzieciom urodzonym z zespołem Beckwitha-Wiedemanna w Stanach Zjednoczonych Ameryki rocznie.
Zespół Lachiewicza-Sibleya
Po raz pierwszy opisany w 1985 roku przez Lachiewicza i wsp. Zespół jest bardzo podobny do Melnick-Fraser, chociaż tylko PAuS i hipoplastyczne nerki z białkomoczem o wczesnym początku zostały znalezione u potomków brytyjskich i irlandzkich imigrantów osiedlających się w Ohio w 1800 roku, a później w Nebrasce. W czasie przeprowadzania oryginalnych badań, spośród 130 żyjących krewnych, 12 członków miało PAuS i hipoplastyczne nerki, 10 miało tylko hipoplastyczne nerki, a 3 miało tylko PAuS . Chociaż dokładne miejsce mutacji pozostaje nieznane, stan ten jest dziedziczony w sposób autosomalny dominujący.
Dwudziesty pierwszy wiek
Dzisiaj, chociaż nadal stosunkowo słabo zbadany, PAuS jest powszechną jednostką kliniczną z dobrze zdefiniowanymi strategiami klinicznymi leczenia. Niektóre badania zidentyfikowały genetyczne powiązanie między PAuS a locus 8q11.1-13.1; jednak wyniki te nie zostały szeroko odtworzone. Poza tym pojedynczym badaniem, żadne inne badania nie koncentrowały się na genetycznej przyczynie PAuS ani nie wiązały go z wyżej wymienionymi zespołami genetycznymi, a badania genealogiczne są nieliczne i bardzo rzadkie
Przebieg kliniczny został dobrze opisany, a strategie leczenia, w tym głównie chirurgiczne wycięcie, zostały dobrze ugruntowane w nowoczesnej otorynolaryngologii oraz chirurgii głowy i szyi. Obecnie pacjenci z PAuS, nie związani z zespołem genetycznym, przechodzą bezpieczny proces interwencji z niewielką liczbą opisanych powikłań, podczas gdy pacjenci z zespołami genetycznymi są poddawani tym samym metodom leczenia, a wycięcie PauS nie wpływa na ich ogólny przebieg kliniczny .
.