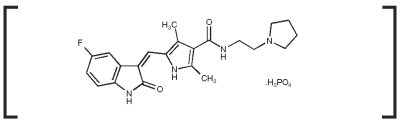
Palladia Indikace
PALLADIA tablety jsou indikovány k léčbě Patnaik II. nebo III. stupně, recidivujících, kožních mastocytárních nádorů s postižením nebo bez postižení regionálních lymfatických uzlin u psů.
Dávkování a způsob podání
K receptu vždy přiložte informační list pro klienta. Podávejte počáteční dávku 3,25 mg/kg (1,48 mg/lb) tělesné hmotnosti, perorálně každý druhý den (viz tabulka 1). Snížení dávky o 0,5 mg/kg (na minimální dávku 2,2 mg/kg (1,0 mg/lb) každý druhý den) a přerušení dávky (přerušení podávání přípravku PALLADIA až na dva týdny) lze v případě potřeby využít ke zvládnutí nežádoucích účinků (viz Tabulka 2 a také Upozornění a opatření). Dávku upravujte na základě přibližně týdenního veterinárního hodnocení po dobu prvních 6 týdnů a poté přibližně každých 6 týdnů. Přípravek PALLADIA lze podávat s krmivem nebo bez něj. Tablety nedělit.
Tabulka 1. 3.Tabulka dávkování 25 mg/kg
|
Tělesná hmotnost psa |
Počet tablet |
|||||
|
Kilogramy |
Kilogramů |
Dávek |
10 mg |
15 mg |
50 mg |
|
|
11.0 – 11,8 |
5,0 – 5,3 |
15 mg |
||||
|
11.9 – 15,2 |
5,4 – 6,9 |
20 mg |
||||
|
15.3 – 18,5 |
7,0 – 8,4 |
25 mg |
||||
|
18.6 – 22,0 |
8,5 – 10,0 |
30 mg |
||||
|
22.1 – 25,4 |
10,1 – 11,5 |
35 mg |
||||
|
25.5 – 28,7 |
11,6 – 13,0 |
40 mg |
||||
|
28.8 – 32,2 |
13,1 – 14,6 |
45 mg |
||||
|
32.3 – 35,5 |
14,7 – 16,1 |
50 mg |
||||
|
35.6 – 38,8 |
16,2 – 17,6 |
55 mg |
||||
|
38.9 – 42,3 |
17,7 – 19,2 |
60 mg |
||||
|
42,4 – 45.6 |
19,3 – 20,7 |
65 mg |
||||
|
45,7 – 50.7 |
20,8 – 23,0 |
70 mg |
||||
|
50,8 – 59,3 |
23.1 – 26,9 |
80 mg |
||||
|
59,4 – 65,9 |
27.0 – 29,9 |
95 mg |
||||
|
66,0 – 71,2 |
30,0 – 32.3 |
100 mg |
||||
|
71,3 – 76,3 |
32,4 – 34.6 |
110 mg |
||||
|
76,4 – 79,6 |
34,7 – 36.1 |
115 mg |
||||
|
79,7 – 84,7 |
36,2 – 38.4 |
120 mg |
||||
|
84,8 – 94.8 |
38,5 – 43,0 |
130 mg |
||||
|
94.9 – 105,0 |
43,1 – 47,6 |
150 mg |
||||
|
105.1 – 110,0 |
47,7 – 49,9 |
160 mg |
||||
|
110.1 – 113,5 |
50,0 – 51,5 |
165 mg |
||||
|
113.6 – 118,6 |
51,6 – 53,8 |
170 mg |
||||
|
118.7 – 128,8 |
53,9 – 58,4 |
180 mg |
||||
|
128.9 – 138,9 |
58,5 – 63,0 |
200 mg |
||||
|
139.0 – 144,0 |
63,1 – 65,3 |
210 mg |
||||
|
144.1 – 157,6 |
65,4 – 71,5 |
215 mg |
||||
|
157.7 – 173,1 |
71,6 – 78,5 |
250 mg |
||||
|
173.2 – 177,9 |
78,6 – 80,7 |
260 mg |
||||
|
178.0 – 191,6 |
80,8 – 86,9 |
265 mg |
||||
|
191.7 – 220,5 |
87,0 – 100,0 |
300 mg |
||||
Tabulka č. 2: Úprava dávky na základě pozorované toxicity
|
Toxicita |
Úprava dávky |
|
Neutropenie |
|
|
>1000/µL |
Udržování hladiny dávky |
|
≤1000/µL nebo neutropenická horečka nebo infekce |
Přestat podávat lék, dokud >1000/µL a klinické příznaky nejsou normální; pak snižte dávku o 0.5 mg/kg |
|
Břišní toxicita (kreatinin) |
|
|
<2.0 mg/dl |
Udržujte hladinu dávky |
|
≥2,0 mg/dl |
Ukončete léčbu, dokud <2.0 mg/dl, pak snižte dávku o 0,5 mg/kg |
|
Albumin |
|
|
<1,5 g/dl |
Ukončete podávání léku do >2.5 g/dl, pak snížit dávku o 0,5 mg/kg |
|
Hematokrit |
|
|
<26% |
Přestat podávat lék do >30%, pak snížit dávku o 0.5 mg/kg |
|
Průjem |
|
|
<4 vodnaté stolice/den po dobu kratší než 2 dny |
Udržovat výši dávky a zahájit podpůrnou léčbu |
|
≥4 vodnaté stolice/den nebo ≥ 2 dny |
Přerušit podávání léku do vytvoření stolice a zahájit podpůrnou léčbu. Při obnovení podávání snižte dávku o 0,5 mg/kg |
|
GI Krvácení |
|
|
Čerstvá krev ve stolici nebo černá dehtovitá stolice po dobu > 2 dnů nebo otevřené krvácení nebo krevní sraženiny ve stolici. |
Ukončení léčby a zahájení podpůrné péče do vymizení všech klinických příznaků krve ve stolici, poté snížení dávky o 0.5 mg/kg. |
Kontraindikace
Nepoužívejte u psů používaných k chovu nebo u březích či laktujících fen (viz klinická farmakologie).
Upozornění
PALLADIA může způsobit vaskulární dysfunkci, která může vést k edému a tromboembolii, včetně plicní tromboembolie. Přerušte léčbu, dokud se klinické příznaky a klinická patologie nenormalizují. Aby byla zajištěna homeostáza cév, vyčkejte alespoň 3 dny po ukončení léčby před provedením chirurgického zákroku (viz Nežádoucí účinky).
U psů léčených přípravkem PALLADIA se vzácně vyskytly závažné a někdy fatální gastrointestinální komplikace včetně gastrointestinální perforace (viz Nežádoucí účinky). Při podezření na gastrointestinální ulcerace přerušte podávání léku a proveďte odpovídající léčbu.
Upozornění pro člověka:
NENÍ URČENO K POUŽITÍ U ČLOVĚKA. UCHOVÁVEJTE TENTO PŘÍPRAVEK A VŠECHNY LÉKY MIMO DOSAH DĚTÍ. Děti by neměly přijít do kontaktu s přípravkem PALLADIA. Chraňte děti před výkaly, močí nebo zvratky léčených psů. Abyste zabránili expozici léku, umyjte si po podání přípravku PALLADIA ruce vodou a mýdlem a noste ochranné rukavice, abyste zabránili přímému kontaktu s výkaly, močí, zvratky a rozlomenými nebo navlhčenými tabletami přípravku PALLADIA. Před obecnou likvidací vložte veškerý odpadní materiál do plastového sáčku a uzavřete jej. Při náhodném zasažení očí přípravkem okamžitě oči vypláchněte vodou. V případě náhodného požití přípravku člověkem okamžitě vyhledejte lékařskou pomoc, lékaři ukažte příbalovou informaci nebo etiketu. Při náhodném požití tohoto přípravku se mohou objevit gastrointestinální potíže, jako je zvracení nebo průjem.
Těhotné ženy, ženy, které mohou otěhotnět, nebo kojící matky by měly věnovat zvláštní pozornost těmto opatřením pro zacházení s přípravkem. (Viz pokyny pro zacházení výše.) Přípravek PALLADIA, stejně jako ostatní léky této skupiny, zabraňuje tvorbě nových cév v nádorech. Podobným způsobem může přípravek PALLADIA ovlivnit tvorbu krevních cév u vyvíjejícího se plodu a může poškodit nenarozené dítě (způsobit vrozené vady). U těhotných žen může mít náhodné požití přípravku PALLADIA nežádoucí účinky na těhotenství.
Upozornění
Pokud se současně objeví anémie, azotémie, hypoalbuminémie a hyperfosfatemie, okamžitě přerušte užívání přípravku PALLADIA. Léčbu obnovte při snížení dávky o 0,5 mg/kg po 1 až 2 týdnech, kdy se hodnoty zlepší a albumin je >2,5 g/dl. Dočasné přerušení léčby může být nutné, pokud se vyskytne některá z těchto hodnot samostatně: hematokrit <26 %, kreatinin ≥2,0 mg/dl nebo albumin <1,5 g/dl. Poté obnovte léčbu při snížení dávky o 0,5 mg/kg, jakmile je hematokrit >30 %, kreatinin <2,0 mg/dl a albumin >2,5 g/dl.
Dočasně přerušte používání přípravku PALLADIA, pokud je počet neutrofilů ≤1000/µl. Léčbu obnovte po 1 až 2 týdnech při snížení dávky o 0,5 mg/kg, když se počet neutrofilů vrátí na >1000/µL. Další snížení dávky může být nutné, pokud se znovu objeví závažná neutropenie.
Přítomnost systémového nádoru žírných buněk před léčbou může psa při léčbě přípravkem PALLADIA predisponovat ke klinicky významné degranulaci žírných buněk s možnými závažnými systémovými nežádoucími reakcemi. Před zahájením léčby přípravkem PALLADIA je třeba se pokusit vyloučit systémovou mastocytózu.
PALLADIA byla spojena s těžkým průjmem nebo krvácením z trávicího traktu, které vyžadují okamžitou léčbu. V závislosti na závažnosti klinických příznaků může být nutné přerušení a snížení dávky. (Viz tabulka 2 v části Dávkování a způsob podání.)
Ve spojení s přípravkem PALLADIA používejte nesteroidní protizánětlivé léky s opatrností kvůli zvýšenému riziku gastrointestinální ulcerace nebo perforace.
PALLADIA se metabolizuje v játrech. Současné podávání přípravku PALLADIA se silnými inhibitory skupiny CYP3A4 může zvýšit koncentrace přípravku PALLADIA. Vliv souběžně podávaných léků, které mohou inhibovat metabolismus přípravku PALLADIA, nebyl hodnocen. U pacientů vyžadujících souběžnou léčbu je třeba sledovat kompatibilitu s léky.
Bezpečné použití přípravku PALLADIA nebylo hodnoceno u psů mladších 24 měsíců nebo vážících méně než 5 kg.
Nežádoucí účinky
V americké klinické terénní studii sestávající z 6týdenní maskované fáze, po které následovala otevřená fáze, byla hodnocena bezpečnost a účinnost přípravku PALLADIA u 151 psů ve vlastnictví klientů, kteří měli Patnaik II. nebo III. stupně, recidivující, kožní mastocytární nádory s postižením regionálních lymfatických uzlin nebo bez něj. Nejčastější nežádoucí účinky hlášené během maskované fáze jsou shrnuty v tabulce 3; nežádoucí účinky hlášené během celé studie (maskovaná fáze kombinovaná s otevřenou fází) jsou shrnuty v tabulce 4.
Tabulka 3. Souhrn nejčastějších nežádoucích účinků během maskované fázea
|
Placebo (n = 64) |
PALLADIA (n = 87) |
|||
|
Nežádoucí účinky. Reakce |
Jakýkoli stupeňb |
Stupeň 3 nebo 4b |
Jakýkoli stupeňb |
Stupeň 3 nebo 4b |
|
Průjem |
26.6% |
3.1% |
46.0% |
6,9% |
|
Anorexie |
31,3% |
6.3% |
39,1% |
6,9% |
|
Letargie |
29.7% |
3.1% |
35.6% |
4.6% |
|
Zvracení |
32,8% |
6,3% |
32,2% |
9.2% |
|
Lameness |
9,4% |
0,0% |
17.2% |
0,0% |
|
Úbytek hmotnosti |
3,1% |
0.0% |
14,9% |
1,1% |
|
Krvácení ve stolici/krvácení z GI/hemoragický průjem |
3.1% |
0.0% |
12.6% |
2.3% |
|
Porucha pohybového aparátu |
6,3% |
0.0% |
11,5% |
1,1% |
|
Dehydratace |
4.7% |
0.0% |
9.2% |
2.3% |
|
Dermatitida |
9,4% |
1,6% |
9,2% |
0.0% |
|
Pruritus |
4,7% |
0,0% |
9.2% |
0,0% |
|
Tachypnoe |
4.7% |
0,0% |
8,0% |
1,1% |
|
Lokalizovaná bolest |
4,7% |
0,0% |
8.0% |
0,0% |
|
Nausea |
3,1% |
0.0% |
8,0% |
1,1% |
|
Celkové bolesti |
4.7% |
1,6% |
6,9% |
0,0% |
|
Polydipsie |
7,8% |
0.0% |
6,9% |
0,0% |
|
Pyrexie |
3.1% |
0.0% |
5.7% |
2.3% |
|
Flatulence |
3,1% |
0,0% |
5.7% |
0,0% |
|
Pigmentační porucha |
1.6% |
0.0% |
5.7% |
0.0% |
|
Laboratorní abnormalita |
Jakýkoli stupeň |
Stupeň 3 nebo 4c |
Jakýkoli Gradec |
Stupeň 3 nebo 4c |
|
Neutropenie |
6.3% |
0.0% |
46.0% |
0,0% |
|
Trombocytopenie |
20,3% |
0,0% |
24,1% |
0.0% |
|
Zvýšená alaninaminotransferáza |
21,9% |
4.7% |
24,1% |
1,1% |
|
Hypoalbuminémie |
7.8% |
0,0% |
12,6% |
0,0% |
|
Snížený hematokrit |
7,8% |
0.0% |
5,7% |
3,4% |
|
Hyperbilirubinémie |
1.6% |
1.6% |
5.7% |
0.0% |
|
Zvýšený kreatinin |
4,7% |
0,0% |
5.7% |
0,0% |
|
Infekce močových cest |
1.6% |
0,0% |
5,7% |
0,0% |
a Průměrná doba trvání studie během maskované fáze byla u psů léčených přípravkem PALLADIA 37,0 dní (medián, 42.0 dní) a 27,6 dní pro psy léčené placebem (medián, 21,0 dní); při statistických srovnáních nebyly provedeny žádné úpravy pro tento rozdíl.
b Zkoušející přidělili stupeň závažnosti 1, 2, 3 nebo 4 (1 – nejméně závažný; 4 – nejzávažnější).
c Známkování laboratorních abnormalit vycházelo ze směrnice National Cancer Institute’s Common Toxicity Criteria upravené pro psy (1 – nejméně závažná; 4 – nejzávažnější).
Tabulka 4. Souhrn nejčastějších nežádoucích účinků během studie (maskovaná fáze v kombinaci s otevřenou fází).label fází)a
|
PALLADIA (n = 145) a |
||
|
Nežádoucí reakce |
Jakýkoli stupeňb |
Stupeň 3 nebo 4b |
|
Průjem |
58.6% |
8,3% |
|
Anorexie |
49.7% |
8,3% |
|
Zvracení |
47.6% |
9,7% |
|
Letargie |
39,3% |
4.1% |
|
Lamita |
22,8% |
0,0% |
|
Ztráta hmotnosti |
21,4% |
2.8% |
|
Krvácení ve stolici/krvácení z GI/hemoragický průjem |
18.6% |
2,8% |
|
Dehydratace |
15,2% |
2.1% |
|
Pruritus |
12,4% |
0,0% |
|
Pigmentační porucha |
11.7% |
0,0% |
|
Dermatitida |
11.0% |
0,0% |
|
Porucha pohybového aparátu |
11,0% |
0,0% |
|
Celkové bolesti |
8.3% |
0,0% |
|
Otitis externa |
8.3% |
0,0% |
|
Tachypnoe |
8,3% |
0.0% |
|
Nausea |
7,6% |
1.4% |
|
Polydipsie |
7,6% |
0,0% |
|
Pyrexie |
6.9% |
2,8% |
|
Arthritis |
6,2% |
0,0% |
|
Lokalizovaný edém |
6.2% |
0,0% |
|
Bakteriální infekce kůže |
5,5% |
0,0% |
|
Zánět spojivek |
5.5% |
0.0% |
|
Laboratorní abnormalita |
Jakýkoli Gradec |
Stupeň 3 nebo 4c |
|
Neutropenie |
44.8% |
1,4% |
|
Hypoalbuminémie |
28,3% |
1.4% |
|
Trombocytopenie |
28,3% |
2.1% |
|
Zvýšená alaninaminotransferáza |
27,6% |
4.1% |
|
Snížený hematokrit |
11,0% |
2.8% |
|
Zvýšený kreatinin |
13,8% |
1,4% |
|
Hyperbilirubinémie |
6,9% |
0.0% |
|
Infekce močových cest |
7,6% |
0,0% |
a Délka léčby přípravkem PALLADIA se pohybovala od 2 do 812 dnů (průměr 144 dnů; medián 68 dnů). Všichni psi dostali alespoň 1 dávku přípravku PALLADIA.
b Zkoušející přidělili stupeň závažnosti 1, 2, 3 nebo 4 (1 – nejméně závažný; 4 – nejzávažnější).
c Hodnocení laboratorních abnormalit vycházelo ze směrnice National Cancer Institute’s Common Toxicity Criteria upravené pro psy (1 – nejméně závažný; 4 – nejzávažnější).
Byly hlášeny další nežádoucí účinky, které se však vyskytly u <5 % psů. Každý jednotlivý pes mohl mít více nežádoucích příhod.
Během této studie došlo k 5 úmrtím, která pravděpodobně souvisela s léčivem. Patologické nálezy obecně odhalily známky vaskulární dysfunkce včetně plicní tromboembolie (pooperační); multiorgánové selhání spojené s vaskulitidou a trombózou; vaskulární trombóza s diseminovanou intravaskulární koagulopatií (DIC) a pankreatitida; a vaskulitida s DIC. Jeden pes uhynul sekundárně na perforaci žaludku; doba léčby přípravkem PALLADIA byla 221 dní a při nekropsii nebyl prokázán tumor žírných buněk. K těmto úmrtím došlo v přítomnosti nebo nepřítomnosti hrubého onemocnění; délka léčby se pohybovala od 18 do 221 dnů.
Souvislost následujících úmrtí s léčivem není známa. Jeden pes, který byl nejprve 3 týdny léčen placebem, uhynul z neznámé příčiny 7 dní po zahájení léčby přípravkem PALLADIA. Další pes uhynul z neznámé příčiny 92 dní po zahájení léčby přípravkem PALLADIA. U žádného z těchto psů nebyla provedena nekropsie.
U 27 psů došlo k nějaké formě gastrointestinálního krvácení, přičemž u 2,8 % psů bylo krvácení závažné. U jednoho psa se objevila žaludeční ulcerace, která pravděpodobně souvisela s lékem. Tři psi během studie zemřeli na perforaci žaludku (1 pes) nebo dvanáctníku (2 psi). Jeden pes s perforací dvanáctníku dostal pouze 1 dávku studovaného léku, a proto nebyl považován za související s lékem.
U sedmi psů se během prvních několika týdnů léčby objevila depigmentace nosu. U jedenácti psů se během studie objevily změny barvy srsti nebo kůže. U dvou z těchto psů došlo k úplným změnám barvy srsti z plavé na bílou a ze sytě červené na blond. U sedmi psů se vyskytla alopecie.
Vliv léku na tělesnou hmotnost: 20,0 % psů mělo >13% úbytek hmotnosti v maskované plus otevřené fázi přičitatelný léku. Z toho 5 psů mělo >25% úbytek hmotnosti.
Tři psi měli během užívání studovaného léku aktivitu podobnou záchvatům. Nelze určit, zda souvisely s lékem.
U dvou psů se objevila epistaxe, která nesouvisela s trombocytopenií. U dalšího psa se vyvinula epistaxe se současnou diseminovanou intravaskulární koagulopatií.
Pro kopii bezpečnostního listu (SDS) nebo pro hlášení nežádoucích účinků volejte společnost Zoetis na číslo 1-888-963-8471.
Informace pro majitele psů:
Vždy spolu s receptem poskytněte informační list pro klienta a prostudujte jej s majiteli. Majitelé by měli být informováni o možných nežádoucích účincích a o tom, kdy je třeba lék vysadit a zavolat veterinárního lékaře. Majitelé by měli být informováni o pokynech pro zacházení s přípravkem.
Klinická farmakologie
Mechanismus účinku: Fosfát toceranibu je malá molekula, která má přímou protinádorovou i antiangiogenní aktivitu. V neklinických farmakologických studiích toceranib selektivně inhiboval tyrozinkinázovou aktivitu několika členů rozštěpené rodiny receptorových tyrozinkináz (RTK), z nichž některé se podílejí na růstu nádorů, patologické angiogenezi a metastatické progresi rakoviny. Toceranib inhiboval aktivitu tyrozinkinázy Flk-1/KDR (receptor pro vaskulární endoteliální růstový faktor, VEGFR2), receptoru pro destičkový růstový faktor (PDGFR) a receptoru pro faktor kmenových buněk (Kit) v biochemických i buněčných testech. Bylo prokázáno, že toceranib působí antiproliferačně na endoteliální buňky in vitro. Léčba toceranibem může vyvolat zástavu buněčného cyklu a následnou apoptózu u nádorových buněčných linií exprimujících aktivační mutace v rozštěpené kináze RTK, ckit. Růst psích žírných buněk je často poháněn aktivačními mutacemi v c-kit.1,2
Jiné sloučeniny ze skupiny antiangiogenetických antineoplastických látek jsou známé tím, že zvyšují embryoletalitu a abnormality plodu. Vzhledem k tomu, že angiogeneze je kritickou složkou embryonálního a fetálního vývoje, je třeba očekávat, že inhibice angiogeneze po podání přípravku PALLADIA bude mít za následek nežádoucí účinky na těhotenství feny.
Farmakokinetika
Po intravenózním podání je farmakokinetika toceranibu charakterizována velmi velkým distribučním objemem (>20 l/kg, což ukazuje na rozdělení do tkání), terminálním eliminačním poločasem přibližně 16 hodin a clearance >1 l/h/kg. Při režimu dávek 3,25 mg ekvivalentu volné báze (fbe)/kg toceranibu podávaných tabletami perorálně každý druhý den po dobu 2 týdnů (7 dávek) jsou farmakokinetické parametry toceranibu v plazmě u zdravých psů plemene bígl (mezi 7,2 – 12,5 kg) uvedeny v následující tabulce.
Tabulka 5: Farmakokinetické parametry
|
Farmakokinetické parametry (průměr + 1SD) |
Celkem (n=11;6M, 5F) |
Celkem (n=10; 5M, 5F) |
|
Eliminační poločas, t1/2 (h) |
16.4 ± 3,6 |
17,2 ± 3,9 |
|
Čas do dosažení maximální plazmatické koncentrace, Tmax (h) |
5.3 ± 1.6 |
6.2 ± 2.6 |
|
Maximální plazmatická koncentrace, Cmax (ng/ml) |
86 ± 22 |
109 ± 41 |
|
Cmin (ng/ml) a, b |
12.7 ± 6.0 |
18.7 ± 8.3 |
|
Plocha pod časovou křivkou plazmatické koncentrace, AUC0-48 (ng-h/ml) a |
1833 ± 508 |
2635 ± 939 |
a Normalizovaná hodnota dávky (upravená na 3.25 mg/kg dávky)
b Cmin je koncentrace po 48 hodinách po podání dávky, což odpovídá dávkovacímu intervalu.
Orální biologická dostupnost toceranibu je 77 %. PALLADIA je vysoce vázána na bílkoviny z 91 % až 93 %.
Je třeba poznamenat, že navzdory homogenitě subjektů zahrnutých do této studie byla pozorována velká variabilita mezi subjekty. Bez ohledu na způsob podání byla při dávkách do 5 mg/kg dvakrát denně pozorována lineární farmakokinetika. Pomocí testovacího systému in vitro na hepatocytech a jaterních mikrosomech bylo zjištěno, že izomer Z je u psů, lidí, koček a potkanů metabolizován na N-oxidový derivát toceranibu. Ačkoli byl ve studii in vitro pozorován malý rozdíl mezi pohlavími (81% konverze u psů, 56% konverze u fen), in vivo nebyly pozorovány žádné rozdíly ve farmakokinetice toceranibu. Vliv poškození ledvin, jater nebo plemene na farmakokinetiku toceranibu nebyl zkoumán.
Účinnost
Účinnost a bezpečnost přípravku PALLADIA perorální tablety pro léčbu mastocytárních nádorů byla hodnocena v randomizované, placebem kontrolované, dvojitě maskované, multicentrické klinické terénní studii. Účelem této studie bylo zhodnotit účinnost a bezpečnost přípravku PALLADIA při léčbě mastocytárních nádorů u psů, u kterých došlo k recidivě měřitelného onemocnění po operaci, a zhodnotit objektivní odpověď (kompletní nebo částečná odpověď). Léčba přípravkem PALLADIA byla porovnávána s léčbou placebem pomocí míry odpovědi na konci 6týdenní maskované fáze. Míra odpovědi byla stanovena pomocí směrnice National Cancer Institute’s Response Evaluation Criteria in Solid Tumors Guideline3 , která byla upravena speciálně pro hodnocení psích mastocytárních nádorů.
Jedno sto padesát tři psů bylo náhodně přiřazeno k léčbě přípravkem PALLADIA v dávce 3,25 mg/kg (n = 88) nebo placebem (n = 65) perorálně, každý druhý den po dobu 6 týdnů nebo do progrese onemocnění nebo do odstoupení ze studie z jiného důvodu. Léčba byla ukončena v době progrese onemocnění: psům, kteří dostávali placebo, byl poté nabídnut přechod na otevřenou léčbu přípravkem PALLADIA; psi, kteří dostávali PALLADIA, byli ze studie vyřazeni. Psi museli mít Patnaikův stupeň II nebo III, recidivující, kožní mastocytární nádory s postižením regionálních lymfatických uzlin nebo bez něj. Alespoň 1 nádor musel mít průměr alespoň 20 mm. Psi měli limit 1 dokončeného protokolu ozařování a limit 1 předchozího režimu systémové chemoterapie. Psi s prokázaným systémovým mastocytomem byli vyloučeni. Léčba systémovými kortikosteroidy během studie nebo během 14 dnů před zahájením studie nebyla povolena. V případě potřeby zvládnutí nežádoucích účinků bylo předepsáno přerušení dávky (přerušení podávání přípravku PALLADIA až na 2 týdny) a/nebo byla dávka snížena až na 2,2 mg/kg.
Analýza účinnosti prokázala statisticky významnou výhodu přípravku PALLADIA oproti placebu v primárním cílovém ukazateli účinnosti, kterým byla objektivní odpověď na konci šestitýdenní maskované fáze. Objektivní odpověď je kompletní odpověď + částečná odpověď. Částečná odpověď je ≥ 30% pokles součtu nejdelších průměrů cílových lézí, přičemž jako referenční hodnota se bere výchozí součet, neprogrese necílových lézí a výskyt žádných nových lézí.
Mastocelulární nádor – výsledky primárního koncového bodu účinnosti
|
Parametr účinnosti |
Placebo (n = 63) |
PALLADIA (n = 86) |
P-.hodnota |
|
Objektivní míra odpovědi * |
7.9% |
37.2% |
< 0.001 |
* Rozdíl v míře objektivní odpovědi mezi skupinami nebyl významně spojen s nádorovou zátěží (přítomnost vs. nepřítomnost postižení regionálních lymfatických uzlin) nebo stupněm nádoru (P > 0,05).
Během studie byl přípravek PALLADIA podáván současně s dalšími léky, jako jsou antimikrobiální přípravky, blokátory H-2 receptorů, antihistaminika, antiemetika, nesteroidní protizánětlivé léky, lokálně působící léky proti vředové chorobě, opiátové modifikátory gastrointestinální motility, opioidy, vakcíny, anthelmintika, antiparazitika a topické/oftalmické/otické kortikosteroidní přípravky. Pouze během otevřené fáze dostalo 5 psů krátkou kúru krátkodobě působících kortikosteroidů.
Bezpečnost u zvířat:
V níže uvedené cílové studii bezpečnosti u zvířat bylo prokázáno, že přípravek PALLADIA má úzké rozpětí bezpečnosti; psi léčení přípravkem PALLADIA by měli být sledováni z hlediska nežádoucích reakcí, které mohou naznačovat nutnost úpravy dávky. Dva psi ve skupině 6 mg/kg byli eutanazováni pro klinickou toxicitu 23. a 27. den studie.
Toceranib byl podáván perorálně 20 samcům a 20 samicím dospělých psů plemene bígl (přibližně 2 roky věku) v dávkách 0 mg/kg (placebo, 12 psů), 2 mg/kg (0,5x, 8 psů), 4 mg/kg (1x, 12 psů) nebo 6 mg/kg (1,5x, 8 psů) jednou denně po dobu 13 po sobě jdoucích týdnů bez přerušení podávání. Toceranib způsobil úbytek hmotnosti, snížení spotřeby krmiva, změny na slinivce břišní, gonádách, nadledvinách, svalech a krvetvorbě.
Spotřeba krmiva se snížila ve skupině 6 mg/kg ve srovnání s placebem, přičemž největší rozdíl v prostředcích se objevil ve 35. dni. Snížení tělesné hmotnosti ve skupině 4 mg/kg bylo pozorováno v 31. den a ve skupině 6 mg/kg v 15. den ve srovnání s placebem a pokračovalo po celou dobu studie. Kulhání související s dávkou, pozorované téměř výhradně u zadních končetin, a bolestivost končetin byla ve všech léčebných skupinách vyšší ve srovnání s placebem, přičemž skupina 6 mg/kg vykazovala nejvyšší výskyt. Ztuhlost a slabost se vyskytovaly téměř výhradně ve skupině 6 mg/kg. Ve všech léčených skupinách bylo pozorováno zarudnutí ústní sliznice. Jeden pes ve skupině 4 mg/kg měl ulcerace v dutině ústní a jeden pes ve skupině 6 mg/kg měl kožní ulcerace, u obou byly přítomny bakteriální infekce. Průjem nebo měkká stolice byly pozorovány ve všech čtyřech skupinách.
Hematologické analýzy ukázaly pokles hematokritu, hemoglobinu a počtu erytrocytů a pokles počtu retikulocytů ve skupinách 4 a 6 mg/kg, které měly tendenci se zotavit natolik, aby omezily další pokles počtu erytrocytů. Počet bílých krvinek byl po celou dobu studie významně nižší ve všech léčených skupinách ve srovnání s placebem, především v důsledku poklesu neutrofilů. Lymfocyty se snížily v menší míře, zejména při nízké dávce. Eosinofily a bazofily vykazovaly výrazný, přetrvávající pokles. Monocyty nebyly ovlivněny.
Počet krevních destiček se mírně zvýšil ve skupinách 4 a 6 mg/kg. Zvýšení bylo pozorováno u fibrinogenu ve skupině 4 a 6 mg/kg.
Zvýšení bylo pozorováno u aspartátaminotransferázy, kreatinkinázy a sérových koncentrací fosforu ve skupinách 4 a 6 mg/kg. Zvýšení alkalické fosfatázy bylo pozorováno ve skupině 6 mg/kg. Zvýšení amylázy bylo pozorováno u jednoho psa v každé ze skupin. Zvýšení sérového draslíku bylo pozorováno u jednoho psa ve skupině 6 mg/kg. Zvýšení laktátdehydrogenázy a globulinů bylo pozorováno ve skupině 6 mg/kg.
Mikroskopické změny související s léčbou zahrnovaly mírné až výrazné snížení buněčnosti hrudní a stehenní kostní dřeně. Ve slezině došlo k odpovídající mírné extramedulární hematopoéze, především erytropoéze. Ve slinivce břišní došlo v souvislosti s dávkou k mírné až středně závažné degranulaci acinárů, charakterizované difuzní ztrátou zymogenních granulí. V nadledvinkách došlo při všech dávkách k minimální kortikální kongesci/krvácení s naznačeným vztahem k dávce. Vakuolizace kůry nadledvin byla zaznamenána s nízkou frekvencí ve všech skupinách. V reprodukčních orgánech obou pohlaví byly zaznamenány změny související s dávkou. U mužů došlo v závislosti na dávce k vyčerpání zárodečných buněk, tubulární vakuolizaci a snížení počtu zralých spermií. U samic vykazovaly vaječníky snížený výskyt zralých/regresujících corpora lutea a zvýšený výskyt malých folikulů.
Dva psi (jeden samec, jedna samice) ve skupině 6 mg/kg byli utraceni pro klinickou toxicitu související s léčbou ve 23. a 27. dni studie. Nástup terminálního syndromu se projevil jako výrazně snížený příjem krmiva a meléna. Během následujících 9 dnů se snížený příjem krmiva rozvinul do téměř úplné anorexie a objevila se hematochezie. Byl pozorován úbytek hmotnosti, letargie, kulhání zadních končetin a slabost. Následující výsledky klinické patologie odpovídají změnám pozorovaným u ostatních psů ve skupině 6 mg/kg a také změnám způsobeným oslabením psů těsně před eutanazií. U obou psů došlo ke zvýšení celkové bílkoviny, globulinů, fosforu, cholesterolu, triglyceridů a fibrinogenu. Jeden pes měl pancytopenii, snížený hematokrit, hemoglobin, retikulocyty, albumin a PT a zvýšené pásy. Přítomna byla také hematurie. Druhý pes měl rovněž snížené lymfocyty, eozinofily, chloridy a sodík a zvýšené hodnoty RBC, hematokritu, hemoglobinu, trombocytů, ALP, amylázy, kreatininu, BUN, hořčíku, draslíku a celkového bilirubinu. Profil srážlivosti ukázal snížení PT a zvýšení PTT u obou psů. Kromě mikroskopických nálezů popsaných u zvířat, která přežila do konce studie, vykazovali tito psi lymfoidní depleci v lymfatických uzlinách, thymu a lymfatických tkáních spojených se střevem a mírné až výrazné gastrointestinální léze. Tito dva psi měli také léze v gastrointestinálním traktu, ledvinách, slinivce břišní, hypofýze a nadledvinách.
Skladovací podmínky:
Jak se přípravek podává
Tablety přípravku PALADIA obsahují 10 mg, 15 mg nebo 50 mg toceranibu ve formě toceranib-fosfátu v jedné tabletě. Tablety jsou baleny v lahvičkách po 30 kusech.
1 London CA, Hannah AL, Zadovoskaya R, et al. Phase I Dose-Escalating Study of SU11654, a Small Molecule Receptor Tyrosine Kinase Inhibitor, in Dogs with Spontaneous Malignancies. Clinical Cancer Research 9(7):2755-2768; 2003
2 Pryer NK, Lee LB, Zadovoskaya R, et al. Proof of Target for SU11654: Inhibition of KIT phosphorylation in Canine Mast Cell Tumors. Clinical Cancer Research 9(15):5729-5734; 2003
3 http://ctep.cancer.gov/protcolDevelopment/
NADA #141-295, Schváleno FDA
Vyrobeno: Pfizer Inc, Ascoli, Itálie
Distribuce: Pfizer Inc:
Revidováno: Zoetis Inc., Kalamazoo, MI 49007
Doplněno: srpen 2015