Molekylær kloning, et begreb, der er kommet til at betyde skabelse af rekombinante DNA-molekyler, har sat skub i udviklingen inden for alle biovidenskaberne. Fra 1970’erne med opdagelsen af restriktionsendonukleaser – enzymer, der selektivt og specifikt skærer DNA-molekyler – har rekombinant DNA-teknologien oplevet en eksponentiel vækst i både anvendelse og raffinement, hvilket har givet stadig kraftigere værktøjer til DNA-manipulation. Kloning af gener er nu så enkel og effektiv, at det er blevet en standardlaboratorieteknik. Dette har ført til en eksplosion i forståelsen af genernes funktion i de seneste årtier. Nye teknologier lover endnu større muligheder, som f.eks. at forskerne kan sammensætte flere DNA-fragmenter og transformere de resulterende plasmider i bakterier på under to timer, eller at der kan anvendes udskiftelige genkassetter, som let kan flyttes mellem forskellige konstruktioner for at maksimere hastigheden og fleksibiliteten. I den nærmeste fremtid vil molekylær kloning sandsynligvis opleve et nyt paradigme med syntetisk biologiske teknikker, som vil gøre det muligt at foretage kemisk in vitro-syntese af enhver in siliko-specificeret DNA-konstruktion. Disse fremskridt bør gøre det muligt at konstruere og gentage DNA-kloner hurtigere og fremskynde udviklingen af genterapivektorer, rekombinante proteinproduktionsprocesser og nye vacciner.
Rebecca Tirabassi, Bitesize Bio.
Indledning
Molekylær kloning henviser til isolering af en DNA-sekvens fra en art (ofte et gen) og dens indsættelse i en vektor med henblik på formering uden ændring af den oprindelige DNA-sekvens. Når molekylære kloner er isoleret, kan de anvendes til at generere mange kopier af DNA’et med henblik på analyse af gensekvensen og/eller til at udtrykke det resulterende protein med henblik på undersøgelse eller udnyttelse af proteinets funktion. Klonerne kan også manipuleres og muteres in vitro for at ændre proteinets ekspression og funktion.
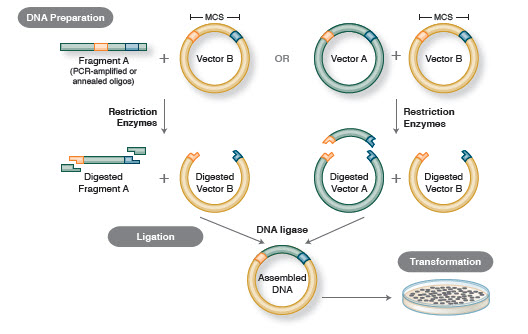
Den grundlæggende arbejdsgang for kloning omfatter fire trin:
- Isolering af mål-DNA-fragmenter (ofte benævnt inserts)
- Ligering af inserts i en passende kloningsvektor, hvorved rekombinante molekyler (f.eks, plasmider)
- Transformation af rekombinante plasmider i bakterier eller andre egnede værter til formering
- Screening/udvælgelse af værter, der indeholder det påtænkte rekombinante plasmid
Disse fire banebrydende trin blev omhyggeligt sat sammen og udført af flere laboratorier, begyndende i slutningen af 1960’erne og begyndelsen af 1970’erne. En oversigt over de opdagelser, der omfatter traditionel molekylær kloning, er beskrevet på de følgende sider.
Historien om kloning
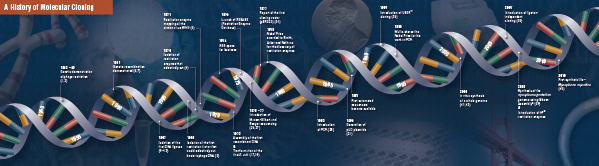
Fundamentet for molekylær kloning
Skæring (fordøjelse). Rekombinant DNA-teknologi opstod først i slutningen af 1960’erne med opdagelsen af enzymer, der specifikt kunne skære og sammenføje dobbeltstrengede DNA-molekyler. Faktisk observerede to grupper allerede i 1952 uafhængigt af hinanden, at bakterier kodede for en “restriktionsfaktor”, som forhindrede bakteriofager i at vokse i visse værter (1,2). Men arten af denne faktor blev først opdaget i 1968, da det lykkedes Arber og Linn at isolere et enzym, kaldet en restriktionsfaktor, som selektivt skar eksogent DNA, men ikke bakterie-DNA (3). Disse undersøgelser identificerede også et methylaseenzym, der beskyttede bakterie-DNA’et mod restriktionsenzymer.
Kort efter Arber og Linns opdagelse udvidede og bekræftede Smith disse undersøgelser ved at isolere et restriktionsenzym fra Haemophilus influenza. Han påviste, at enzymet selektivt skar DNA midt i en specifik 6 basepars DNA-strækning; et karakteristisk træk ved visse restriktionsenzymer er deres tilbøjelighed til at skære DNA-substratet i eller i nærheden af specifikke, ofte palindromiske, “genkendelses “sekvenser (4).
Den fulde styrke af restriktionsenzymer blev først erkendt, da restriktionsenzymer og gelelektroforese blev anvendt til at kortlægge genomet af Simian Virus 40 (SV40) (5). For disse banebrydende resultater delte Werner Arber, Hamilton Smith og Daniel Nathans Nobelprisen i medicin i 1978.
Figur 1. Traditionel arbejdsgang for kloning
Assembling (Ligation). På samme måde som opdagelsen af enzymer, der klipper DNA, gik opdagelsen af et enzym, der kan sammenføje DNA, forud for tidligere, markante observationer. I begyndelsen af 1960’erne opdagede to grupper, at genetisk rekombination kunne finde sted gennem brud på og ligering af DNA-molekyler (6,7), tæt fulgt af observationen af, at lineært bakteriofag-DNA hurtigt omdannes til kovalent lukkede cirkler efter infektion af værten (8). Blot to år senere isolerede fem grupper uafhængigt af hinanden DNA-ligaser og påviste deres evne til at samle to DNA-stykker (9-13).
Snart efter opdagelsen af restriktionsenzymer og DNA-ligaser blev det første rekombinante DNA-molekyle fremstillet. I 1972 skar og ligerede Berg separat et stykke lambda bakteriofag-DNA eller E. coli galaktoseoperonet med SV40-DNA for at skabe de første rekombinante DNA-molekyler (14). Disse undersøgelser var banebrydende for konceptet om, at DNA på grund af DNA’s universelle karakter kunne forbindes med DNA fra alle arter. I 1980 delte Paul Berg Nobelprisen i kemi med Walter Gilbert og Frederick Sanger (udviklerne af DNA-sekventering) for “hans grundlæggende studier af nukleinsyrernes biokemi, især med hensyn til rekombinant DNA”.
Transformation. Rekombinant DNA-teknologi ville være stærkt begrænset, og molekylær kloning ville være umulig uden midler til at formere og isolere det nykonstruerede DNA-molekyle. Evnen til at transformere bakterier eller fremkalde optagelse, inkorporering og ekspression af fremmed genetisk materiale blev først demonstreret af Griffith, da han transformerede en ikke-dødelig bakteriestamme til en dødelig stamme ved at blande den ikke-dødelige stamme med varmeinaktiverede dødelige bakterier (15). Man forstod imidlertid ikke arten af det “transformerende princip”, der gav dødelighed, før man i 1944 forstod, hvad det var for et princip. Samme år påviste Avery, Macleod og McCarty, at det var DNA og ikke protein, der var ansvarlig for at fremkalde den dødelige fænotype (16).
I begyndelsen troede man, at den almindelige bakteriestamme til laboratoriebrug, E. coli, var modstandsdygtig over for transformation, indtil Mandel og Higa påviste, at behandling af E. coli med calciumklorid fremkaldte optagelsen af bakteriofag-DNA (17). Cohen anvendte dette princip i 1972, da han var pioner inden for transformation af bakterier med plasmider med henblik på at give bakterierne antibiotikaresistens (18).
Det ultimative eksperiment: fordøjelse, ligering og transformation af et rekombinant DNA-molekyle blev udført af Boyer, Cohen og Chang i 1973, da de fordøjede plasmidet pSC101 med EcoRI, ligerede det lineariserede fragment til et andet enzymbegrænset plasmid og transformerede det resulterende rekombinante molekyle i E. coli og gav bakterierne tetracyklinresistens (19), hvorved de lagde grunden til det meste arbejde med rekombinant DNA siden da.
Bygger på grundarbejdet
Selv om forskerne havde opdaget og anvendt alle de grundlæggende principper for at skabe og formere rekombinant DNA i bakterier, var processen ineffektiv. Restriktionsenzympræparater var upålidelige på grund af ikke-standardiserede oprensningsprocedurer, plasmider til kloning var besværlige, vanskelige at arbejde med og begrænsede i antal, og eksperimenterne var begrænset af mængden af indsat DNA, der kunne isoleres. Forskning i løbet af de næste par årtier førte til forbedringer af de teknikker og værktøjer, der var til rådighed for molekylær kloning.
Første vektordesign.
Udvikling af den første standardiserede vektor. Forskere, der arbejdede i Boyers laboratorium, erkendte behovet for et generelt kloningsplasmid, et kompakt plasmid med unikke restriktionssteder til kloning af fremmed DNA og ekspression af antibiotikaresistensgener med henblik på selektion af transformerede bakterier. I 1977 beskrev de den første vektor designet til kloningsformål, pBR322 (20). Denne vektor var lille, ~4 kilobaser i størrelse, og havde to antibiotikaresistensgener til selektion.
Vektorer med on-board screening og højere udbytte. Selv om antibiotisk selektion forhindrede ikke-transformerede bakterier i at vokse, kunne plasmider, der re-ligerede uden indsatte DNA-fragmenter (selv-ligering), stadig give bakterier antibiotikaresistens. Derfor kunne det være tidskrævende at finde de korrekte bakteriekloner, der indeholder det ønskede rekombinante DNA-molekyle.
Vieira og Messing udtænkte et screeningsværktøj til at identificere bakteriekolonier, der indeholder plasmider med DNA-inserts. Med udgangspunkt i pBR322-plasmidet skabte de serien af pUC-plasmider, som indeholdt et “blå/hvid-screeningssystem” (21). Ved at placere et multiple kloningssted (MCS), der indeholder flere unikke restriktionssteder inden for LacZ´-genet, kunne forskerne screene bakteriekolonier, der indeholdt plasmider med den fremmede DNA-indsats. Når bakterier blev udplottet på de korrekte medier, indeholdt hvide kolonier plasmider med indsatser, mens blå kolonier indeholdt plasmider uden indsatser. pUC-plasmider havde en yderligere fordel i forhold til eksisterende vektorer; de indeholdt en mutation, der resulterede i et højere antal kopier, hvilket øgede plasmidudbyttet.
Forbedring af restriktionsdigester. Det tidlige arbejde med restriktionsenzymer blev hæmmet af enzympræparatets renhed og manglende forståelse af bufferkravene til de enkelte enzymer. I 1975 blev New England Biolabs (NEB) det første firma til at markedsføre restriktionsenzymer fremstillet fra en rekombinant kilde. Dette gav mulighed for højere udbytte, forbedret renhed, ensartethed fra parti til parti og lavere priser. I øjeblikket er over 4 000 restriktionsenzymer, der genkender over 300 forskellige sekvenser, blevet opdaget af forskere over hele verden . NEB leverer i øjeblikket over 230 af disse specificiteter.
NEB var også en af de første virksomheder til at udvikle et standardiseret firebuffersystem og til at karakterisere alle sine enzymaktiviteter i dette buffersystem. Dette førte til en bedre forståelse af, hvordan man gennemfører en dobbelt fordøjelse, dvs. fordøjelse af DNA med to enzymer samtidig. Senere forskning førte til udvikling af one-buffer-systemer, som er kompatible med de mest almindelige restriktionsenzymer (f.eks. NEB’s CutSmart™ Buffer).
Med fremkomsten af kommercielt tilgængelige restriktionsenzym-biblioteker med kendte sekvensspecificiteter blev restriktionsenzymerne et effektivt redskab til screening af potentielle rekombinante DNA-kloner. Den “diagnostiske digest” var og er stadig en af de mest almindelige teknikker, der anvendes ved molekylær kloning.
Vektor- og insert-præparering. Kloningseffektiviteten og alsidigheden blev også forbedret ved udviklingen af forskellige teknikker til forberedelse af vektorer før ligering. Der blev isoleret alkaliske fosfataser, som kunne fjerne 3´ og 5´ fosfatgrupperne fra DNA’s ender . Man opdagede snart, at behandling af vektorer med kalve-tarmfosfatase (CIP) affosforylerede DNA-enderne og forhindrede selvligering af vektoren, hvilket øgede genfindelsen af plasmider med insert (24).
CIP-enzymet viste sig vanskeligt at inaktivere, og enhver tilbageværende aktivitet førte til affosforylering af insert-DNA og hæmning af ligeringsreaktionen. Opdagelsen af de varmelabile alkaliske fosfataser, såsom rekombinant rejealkalisk fosfatase (rSAP) og antarktisk fosfatase (AP) (begge solgt af NEB), mindskede de involverede trin og den involverede tid, da et simpelt skift i temperatur inaktiverer enzymet før ligeringstrinnet (25).
DNA-sekventering kommer frem. DNA-sekventering blev udviklet i slutningen af 1970’erne, da to konkurrerende metoder blev udtænkt. Maxam og Gilbert udviklede den “kemiske sekventeringsmetode”, som byggede på kemisk modifikation af DNA og efterfølgende spaltning ved specifikke baser (26). Samtidig offentliggjorde Sanger og kolleger den såkaldte “chain-termination-metode”, som blev den metode, der blev anvendt af de fleste forskere (27). Sanger-metoden blev hurtigt automatiseret, og de første automatiske sekvenseringsmaskiner blev solgt i 1987.
Muligheden for at bestemme sekvensen af en DNA-strækning øgede pålideligheden og alsidigheden af molekylær kloning. Når klonerne var klonet, kunne forskerne sekventere klonerne for endeligt at identificere det korrekte rekombinante molekyle, identificere nye gener eller mutationer i gener og nemt designe oligonukleotider baseret på den kendte sekvens til yderligere eksperimenter.
Indflydelse af polymerasekædereaktionen. Et af problemerne ved molekylær kloning i de tidlige år var at skaffe nok insert-DNA til at klone ind i vektoren. I 1983 udtænkte Mullis en teknik, der løste dette problem og revolutionerede den molekylære kloning (28). Han forstærkede en strækning af mål-DNA ved at bruge modsatrettede primere til at forstærke begge komplementære DNA-strenge samtidig. Gennem cyklusser af denaturering, annealing og polymerisering viste han, at han kunne amplificere en enkelt DNA-kopi eksponentielt. Polymerasekædereaktionen, PCR, gjorde det muligt at forstærke og klone gener fra tidligere utilstrækkelige mængder DNA. For denne opdagelse delte Kary Mullis i 1993 Nobelprisen i kemi “for bidrag til udviklingen af metoder inden for DNA-baseret kemi”.
I 1970 opdagede Temin og Baltimore uafhængigt af hinanden omvendt transkriptase i vira, et enzym, der omdanner RNA til DNA (29,30). Kort efter at PCR blev udviklet, blev omvendt transkription koblet med PCR (RT-PCR) for at muliggøre kloning af messenger RNA (mRNA). Omvendt transkription blev anvendt til at skabe en DNA-kopi (cDNA) af mRNA, som efterfølgende blev forstærket ved PCR for at skabe et indstik til ligering. For deres opdagelse af enzymet fik Howard Temin og David Baltimore i 1975 Nobelprisen i medicin og fysiologi, som de delte med Renato Dulbecco.
Kloning af PCR-produkter. Indførelsen af PCR betød, at forskere nu kunne klone gener og DNA-segmenter med begrænset kendskab til amplikonsekvensen. Der var imidlertid kun ringe enighed om den optimale metode til forberedelse af PCR-produkter med henblik på effektiv ligering i kloningsvektorer.
Der blev oprindeligt anvendt flere forskellige metoder til kloning af PCR-produkter. Den enkleste og stadig mest almindelige metode til kloning af PCR-produkter er ved at indføre restriktionssteder i PCR-produktets ender (31). Dette giver mulighed for direkte, retningsbestemt kloning af indsatsen i vektoren efter restriktionsfordøjelsen. Kloning med stumpe ender blev udviklet til direkte ligering af PCR-produkter, der er genereret af polymeraser, der producerer stumpe ender, eller indsatser, der er konstrueret til at have restriktionssteder, der efterlader stumpe ender, når indsatsen er blevet fordøjet. Dette var nyttigt til kloning af DNA-fragmenter, som ikke indeholdt restriktionssteder, der var kompatible med vektoren (32).
Kort efter indførelsen af PCR blev overlap extension PCR introduceret som en metode til at samle PCR-produkter til en sammenhængende DNA-sekvens (33). Ved denne metode amplificeres DNA-indsatsen ved PCR ved hjælp af primere, der genererer et PCR-produkt, som indeholder overlappende regioner med vektoren. Vektoren og indsatsen blandes derefter, denatureres og annealeres, hvilket muliggør hybridisering af indsatsen til vektoren. En anden PCR-runde frembringer rekombinante DNA-molekyler af vektor med insert. Overlap extension PCR har gjort det muligt for forskerne at samle store gener, som ikke let kunne amplificeres ved hjælp af traditionelle PCR-metoder. Overlap extension PCR blev også anvendt til at indføre mutationer i gensekvenser (34).
Figur 2. Oversigt over PCR

Udvikling af specialiserede kloningsteknikker.
I et forsøg på yderligere at forbedre effektiviteten af molekylær kloning blev der udviklet flere specialiserede værktøjer og teknikker, der udnyttede egenskaberne ved unikke enzymer.
TA-kloning. En metode udnyttede en egenskab ved Taq DNA-polymerase, den første varmestabile polymerase, der blev anvendt til PCR. Under amplifikationen tilføjer Taq et enkelt 3´ dA-nukleotid til enden af hvert PCR-produkt. PCR-produktet kan let ligeres ind i en vektor, der er blevet klippet og manipuleret til at indeholde enkelte T-rester på hver streng. Flere firmaer har markedsført denne teknik og sælger kits med kloningsvektorer, der allerede er lineariseret og “tailed”.
LIC. Ligationsuafhængig kloning (LIC) giver, som navnet antyder, mulighed for at sammenføje DNA-molekyler i fravær af DNA-ligase. LIC udføres almindeligvis med T4 DNA-polymerase, som anvendes til at generere enkeltstrengede DNA-overhangs, >12 nukleotider lange, på både det lineariserede vektor-DNA og det indstik, der skal kloneres (35). Når de blandes sammen, annealerer vektoren og indsatsen gennem den lange strækning af kompatible ender. Længden af de kompatible ender er tilstrækkelig lang til at holde molekylet sammen i fravær af ligase, selv under transformationen. Når de er transformeret, repareres hullerne in vivo. Der findes flere forskellige kommercielt tilgængelige produkter til LIC.USER-kloning.
USER-kloning blev først udviklet i begyndelsen af 1990’erne som en restriktionsenzym- og ligaseuafhængig kloningsmetode (36). Da metoden først blev udtænkt, var den baseret på anvendelse af PCR-primere, der indeholdt en ~12 nukleotid 5´-hale, hvor mindst fire deoxythymidinbaser var blevet erstattet med deoxyuridiner. PCR-produktet blev behandlet med uracil DNA-glycosidase (UDG) og endonuklease VIII, som fjerner uracilbaserne og efterlader et 3´overlap, der kan annealeres til en tilsvarende behandlet vektor. NEB sælger USER-enzymet til ligase- og restriktionsenzym-uafhængige kloningsreaktioner.
Fremtidige tendenser
Molekylær kloning har udviklet sig fra kloning af et enkelt DNA-fragment til samling af flere DNA-komponenter i en enkelt sammenhængende DNA-strækning. Nye og nye teknologier søger at omdanne kloning til en proces, der er lige så enkel som at arrangere “blokke” af DNA ved siden af hinanden.
DNA-samlingsmetoder. Mange nye, elegante teknologier gør det muligt at samle flere DNA-fragmenter i en enkelt rørreaktion. Fordelene ved disse teknologier er, at de er standardiserede, problemfri og for det meste sekvensuafhængige. Desuden gør muligheden for at samle flere DNA-fragmenter i ét rør en række tidligere uafhængige restriktions/ligeringsreaktioner til en strømlinet, effektiv procedure.
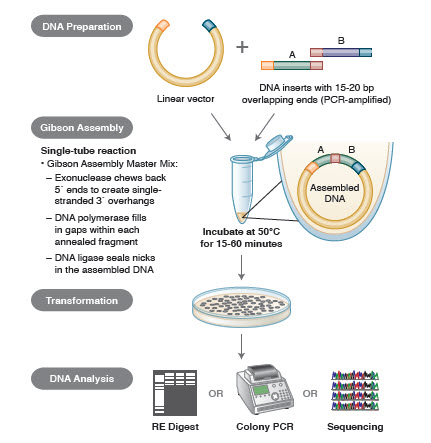
De forskellige teknikker og produkter til genmontering omfatter SLIC (Sequence and Ligase Independent Cloning), Gibson Assembly (NEB), GeneArt® Seamless Cloning (Life Technologies) og Gateway® Cloning (Invitrogen) (35,37,38).
I DNA-samlingen PCR-amplificeres de DNA-blokke, der skal samles. Derefter konstrueres de DNA-fragmenter, der skal samles ved siden af hinanden, således at de indeholder blokke af komplementære sekvenser, der skal ligeres sammen. Det kan være kompatible kohæsive ender, som f.eks. dem, der anvendes til Gibson Assembly, eller regioner, der indeholder genkendelsessteder for stedspecifikke rekombinaser (Gateway). Det enzym, der anvendes til DNA-ligering, genkender og samler hvert sæt af kompatible regioner og skaber et enkelt sammenhængende DNA-molekyle i én reaktion.
Figur 3. Oversigt over Gibson Assembly-kloningsmetoden
Syntetisk biologi. DNA-syntese er et område inden for syntetisk biologi, som i øjeblikket er ved at revolutionere rekombinant DNA-teknologien. Selv om et komplet gen først blev syntetiseret in vitro i 1972 (40), blev DNA-syntese af store DNA-molekyler ikke en realitet før begyndelsen af 2000’erne, hvor forskere begyndte at syntetisere hele genomer in vitro (41,42). Disse tidlige eksperimenter tog år at gennemføre, men teknologien fremskynder evnen til at syntetisere store DNA-molekyler.
Konklusion
I de sidste 40 år har molekylær kloning udviklet sig fra en møjsommelig isolering og sammensætning af to stykker DNA efterfulgt af intensiv screening af potentielle kloner til problemfri samling af op til 10 DNA-fragmenter med bemærkelsesværdig effektivitet på blot få timer eller design af DNA-molekyler in silico og syntese af dem in vitro. Tilsammen giver alle disse teknologier molekylærbiologerne en forbløffende kraftfuld værktøjskasse til at udforske, manipulere og udnytte DNA, hvilket vil udvide videnskabens horisont yderligere. Blandt mulighederne er udvikling af mere sikre rekombinante proteiner til behandling af sygdomme, forbedring af genterapi (43) og hurtigere produktion, validering og frigivelse af nye vacciner (44). Men i sidste ende er potentialet kun begrænset af vores fantasi.
Rebecca Tirabassi er assisterende redaktør på Bitesizebio.com.