Molekylär kloning, en term som har kommit att betyda skapandet av rekombinanta DNA-molekyler, har drivit framstegen inom hela livsvetenskapen. Med början på 1970-talet, med upptäckten av restriktionsendonukleaser – enzymer som selektivt och specifikt skär av DNA-molekyler – har tekniken för rekombinant DNA haft en exponentiell tillväxt både när det gäller tillämpning och sofistikering, vilket har gett upphov till allt kraftfullare verktyg för DNA-manipulering. Kloning av gener är nu så enkel och effektiv att den har blivit en standardlaboratorieteknik. Detta har lett till en explosionsartad ökning av förståelsen av genernas funktion under de senaste årtiondena. Ny teknik lovar ännu större möjligheter, t.ex. att göra det möjligt för forskare att sömlöst sammanfoga flera DNA-fragment och omvandla de resulterande plasmiderna till bakterier på mindre än två timmar, eller att använda utbytbara genkassetter, som lätt kan flyttas mellan olika konstruktioner, för att maximera snabbhet och flexibilitet. Inom en nära framtid kommer den molekylära kloningen troligen att få uppleva ett nytt paradigm med tekniker för syntetisk biologi som kommer att möjliggöra kemisk in vitro-syntes av vilken in silico-specificerad DNA-konstruktion som helst. Dessa framsteg bör möjliggöra snabbare konstruktion och iteration av DNA-kloner, vilket påskyndar utvecklingen av vektorer för genterapi, processer för produktion av rekombinanta proteiner och nya vacciner.
Rebecca Tirabassi, Bitesize Bio.
Introduktion
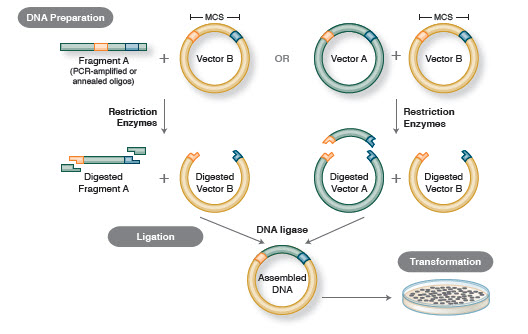
Med molekylär kloning menas isolering av en DNA-sekvens från vilken art som helst (ofta en gen), och dess infogning i en vektor för förökning, utan att den ursprungliga DNA-sekvensen förändras. När de väl har isolerats kan molekylära kloner användas för att generera många kopior av DNA:t för analys av gensekvensen och/eller för att uttrycka det resulterande proteinet för att studera eller utnyttja proteinets funktion. Klonerna kan också manipuleras och muteras in vitro för att ändra proteinets uttryck och funktion.
Det grundläggande arbetsflödet för kloning omfattar fyra steg:
- Isolering av mål-DNA-fragment (ofta kallade inserts)
- Ligering av inserts i en lämplig kloningsvektor, vilket ger upphov till rekombinanta molekyler (t.ex, plasmider)
- Transformation av rekombinanta plasmider i bakterier eller andra lämpliga värddjur för förökning
- Screening/urval av värddjur som innehåller den avsedda rekombinanta plasmiden
Dessa fyra banbrytande steg sattes noggrant ihop och utfördes av flera laboratorier, med början i slutet av 1960-talet och början av 1970-talet. En sammanfattning av de upptäckter som omfattar traditionell molekylär kloning beskrivs på följande sidor.
Historia om kloning
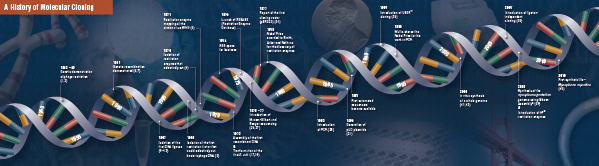
Grunden för molekylär kloning
Skärning (Digestion). Rekombinant DNA-teknik uppstod först i slutet av 1960-talet, när man upptäckte enzymer som specifikt kunde klippa och sammanfoga dubbelsträngade DNA-molekyler. Redan 1952 observerade två grupper oberoende av varandra att bakterier kodade för en ”restriktionsfaktor” som hindrade bakteriofager från att växa i vissa värddjur (1,2). Men faktorens natur upptäcktes inte förrän 1968, då Arber och Linn lyckades isolera ett enzym, kallat restriktionsfaktor, som selektivt klippte exogent DNA, men inte bakteriellt DNA (3). I dessa studier identifierades också ett metylasenzym som skyddade bakteriens DNA från restriktionsenzymer.
Kort efter Arbers och Linns upptäckt utvidgade och bekräftade Smith dessa studier genom att isolera ett restriktionsenzym från Haemophilus influenza. Han visade att enzymet selektivt skar DNA i mitten av en specifik 6 baspars sträcka av DNA; ett kännetecken för vissa restriktionsenzymer är deras benägenhet att skära DNA-substratet i eller nära specifika, ofta palindromiska, ”igenkännings ”sekvenser (4).
Restriktionsenzymernas fulla kraft insågs inte förrän restriktionsenzymer och gelelektrofores användes för att kartlägga genomet av Simian Virus 40 (SV40) (5). För dessa banbrytande upptäckter delade Werner Arber, Hamilton Smith och Daniel Nathans 1978 års Nobelpris i medicin.
Figur 1. Traditionellt arbetsflöde för kloning
Assembling (Ligation). På samma sätt som upptäckten av enzymer som klipper DNA föregicks upptäckten av ett enzym som kan sammanfoga DNA av tidigare, framträdande observationer. I början av 1960-talet upptäckte två grupper att genetisk rekombination kan ske genom att DNA-molekyler bryts och ligeras (6,7), tätt följt av observationen att linjärt bakteriofag-DNA snabbt omvandlas till kovalent slutna cirklar efter infektion av värden (8). Bara två år senare isolerade fem grupper oberoende av varandra DNA-ligaser och visade deras förmåga att sammanfoga två DNA-bitar (9-13).
Snart efter upptäckten av restriktionsenzymer och DNA-ligaser tillverkades den första rekombinanta DNA-molekylen. År 1972 klippte och ligerade Berg separat en bit lambda bakteriofag-DNA eller E. coli galaktosoperon med SV40-DNA för att skapa de första rekombinanta DNA-molekylerna (14). Dessa studier var banbrytande när det gällde konceptet att DNA från alla arter kunde sammanfogas på grund av DNA:s universella karaktär. 1980 delade Paul Berg Nobelpriset i kemi med Walter Gilbert och Frederick Sanger (utvecklare av DNA-sekvensering) för ”hans grundläggande studier av nukleinsyrornas biokemi, med särskild hänsyn till rekombinant DNA”.
Transformation. Rekombinant DNA-teknik skulle vara starkt begränsad, och molekylär kloning omöjlig, utan medel för att föröka och isolera den nykonstruerade DNA-molekylen. Förmågan att transformera bakterier, eller framkalla upptag, införlivande och uttryck av främmande genetiskt material, demonstrerades för första gången av Griffith när han transformerade en icke-dödlig bakteriestam till en dödlig stam genom att blanda den icke-dödliga stammen med värmeinaktiverade dödliga bakterier (15). Man förstod dock inte vad den ”transformerande principen” som gav dödlighet innebar förrän 1944. Samma år visade Avery, Macleod och McCarty att det var DNA, och inte protein, som var ansvarigt för att framkalla den dödliga fenotypen (16).
I början trodde man att den vanliga bakteriestammen E. coli, som används i laboratorier, var refraktär mot omvandling, tills Mandel och Higa visade att behandling av E. coli med kalciumklorid framkallade upptag av bakteriofag-DNA (17). Cohen tillämpade denna princip 1972 när han var pionjär när det gällde att transformera bakterier med plasmider för att ge dem antibiotikaresistens (18).
Det ultimata experimentet: digestion, ligering och transformation av en rekombinant DNA-molekyl utfördes av Boyer, Cohen och Chang 1973, när de digrerade plasmidet pSC101 med EcoRI, ligerade det linjäriserade fragmentet till en annan enzymbegränsad plasmid och transformerade den resulterande rekombinanta molekylen i E. coli och gav bakterien tetracyklinresistens (19) och lade därmed grunden för det mesta arbetet med rekombinant DNA sedan dess.
Bygga vidare på grundarbetet
Sedan dess hade forskarna upptäckt och tillämpat alla grundprinciper för att skapa och föröka rekombinant DNA i bakterier, men processen var ineffektiv. Restriktionsenzymberedningar var opålitliga på grund av icke-standardiserade reningsförfaranden, plasmider för kloning var besvärliga, svåra att arbeta med och begränsade i antal, och experimenten begränsades av mängden insatt DNA som kunde isoleras. Forskningen under de kommande decennierna ledde till förbättringar av de tekniker och verktyg som finns tillgängliga för molekylär kloning.
Första vektordesignen.
Utveckling av den första standardiserade vektorn. Forskare som arbetade i Boyers labb insåg behovet av en allmän kloningsplasmid, en kompakt plasmid med unika restriktionsställen för kloning av främmande DNA och uttryck av antibiotikaresistensgener för selektion av transformerade bakterier. År 1977 beskrev de den första vektorn avsedd för kloning, pBR322 (20). Denna vektor var liten, ~4 kilobaser i storlek, och hade två antibiotikaresistensgener för selektion.
Vektorer med screening ombord och högre avkastning. Även om antibiotikaselektion hindrade icke-transformerade bakterier från att växa, kunde plasmider som återligerade utan insatta DNA-fragment (självligering) fortfarande ge bakterier antibiotikaresistens. Att hitta rätt bakteriekloner som innehåller den önskade rekombinanta DNA-molekylen kunde därför vara tidskrävande.
Vieira och Messing utarbetade ett screeningverktyg för att identifiera bakteriekolonier som innehåller plasmider med DNA-insatser. Med utgångspunkt i pBR322-plasmiden skapade de serien av pUC-plasmider, som innehöll ett ”blå/vit screening”-system (21). Genom att placera en multipel kloneringsplats (MCS) som innehåller flera unika restriktionsplatser inom LacZ´-genen kunde forskarna screena bakteriekolonier som innehöll plasmider med det främmande DNA-inslaget. När bakterier plöjdes ut på rätt media innehöll vita kolonier plasmider med inlagor, medan blå kolonier innehöll plasmider utan inlagor. pUC-plasmiderna hade ytterligare en fördel jämfört med befintliga vektorer; de innehöll en mutation som resulterade i ett högre antal kopior, vilket därför ökade plasmidutbytet.
Förbättring av restriktionsavdrifterna. Det tidiga arbetet med restriktionsenzymer hindrades av enzympreparatets renhet och bristande förståelse för buffertkraven för varje enzym. År 1975 blev New England Biolabs (NEB) det första företaget som kommersialiserade restriktionsenzymer producerade från en rekombinant källa. Detta möjliggjorde högre avkastning, förbättrad renhet, jämnhet från parti till parti och lägre priser. För närvarande har över 4 000 restriktionsenzymer, som känner igen över 300 olika sekvenser, upptäckts av forskare över hela världen. NEB tillhandahåller för närvarande över 230 av dessa specificiteter.
NEB var också ett av de första företagen som utvecklade ett standardiserat system med fyra buffertar och som karakteriserade alla sina enzymaktiviteter i detta buffertsystem. Detta ledde till en bättre förståelse för hur man genomför en dubbel digestion, eller digestion av DNA med två enzymer samtidigt. Senare forskning ledde till utvecklingen av system med en buffert, som är kompatibla med de vanligaste restriktionsenzymerna (t.ex. NEB:s CutSmart™ Buffer).
Med tillkomsten av kommersiellt tillgängliga restriktionsenzymbibliotek med kända sekvensspecificiteter blev restriktionsenzymerna ett kraftfullt verktyg för screening av potentiella rekombinanta DNA-kloner. Den ”diagnostiska digesten” var, och är fortfarande, en av de vanligaste teknikerna som används vid molekylär kloning.
Vektor- och insertberedning. Kloningens effektivitet och mångsidighet förbättrades också genom utvecklingen av olika tekniker för att förbereda vektorer före ligering. Alkaliska fosfataser isolerades som kunde avlägsna 3´ och 5´ fosfatgrupperna från DNA:s ändar . Man upptäckte snart att behandling av vektorer med kalv-tarmfosfatas (CIP) avfosforylerade DNA-ändarna och förhindrade självligering av vektorn, vilket ökade återvinningen av plasmider med insert (24).
CIP-enzymet visade sig vara svårt att inaktivera, och all kvarvarande aktivitet ledde till avfosforylering av insert-DNA och hämning av ligeringsreaktionen. Upptäckten av de värmelabila alkaliska fosfataserna, såsom rekombinant Shrimp Alkaline Phosphatase (rSAP) och Antarctic Phosphatase (AP) (båda säljs av NEB), minskade stegen och tiden, eftersom en enkel temperaturförskjutning inaktiverar enzymet före ligeringssteget (25).
DNA-sekvenseringen anländer. DNA-sekvensering utvecklades i slutet av 1970-talet när två konkurrerande metoder utarbetades. Maxam och Gilbert utvecklade den ”kemiska sekvenseringsmetoden”, som byggde på kemisk modifiering av DNA och efterföljande klyvning vid specifika baser (26). Samtidigt publicerade Sanger och kollegor ”chain-termination-metoden”, som blev den metod som användes av de flesta forskare (27). Sangermetoden blev snabbt automatiserad, och de första automatiska sekvenseringsapparaterna såldes 1987.
Möjligheten att bestämma sekvensen för en DNA-sträcka ökade tillförlitligheten och mångsidigheten för molekylär kloning. När kloningen väl var klar kunde forskarna sekvensera klonerna för att slutgiltigt identifiera den korrekta rekombinanta molekylen, identifiera nya gener eller mutationer i gener och enkelt utforma oligonukleotider baserade på den kända sekvensen för ytterligare experiment.
Polymeraskedjereaktionens inverkan. Ett av problemen med molekylär kloning under de första åren var att få fram tillräckligt med insatt DNA för att klona in i vektorn. År 1983 utarbetade Mullis en teknik som löste detta problem och revolutionerade den molekylära kloningen (28). Han amplifierade en sträcka av mål-DNA genom att använda motsatta primers för att amplifiera båda de komplementära DNA-strängarna samtidigt. Genom cykler av denaturering, glödgning och polymerisering visade han att han exponentiellt kunde amplifiera en enda kopia av DNA. Polymeraskedjereaktionen, PCR, gjorde det möjligt att amplifiera och klona gener från tidigare otillräckliga mängder DNA. För denna upptäckt delade Kary Mullis 1993 Nobelpriset i kemi ”för bidrag till utvecklingen av metoder inom DNA-baserad kemi”.
1970 upptäckte Temin och Baltimore oberoende av varandra omvänt transkriptas i virus, ett enzym som omvandlar RNA till DNA (29,30). Kort efter att PCR utvecklats kopplades omvänd transkription till PCR (RT-PCR) för att möjliggöra kloning av messenger RNA (mRNA). Omvänd transkription användes för att skapa en DNA-kopia (cDNA) av mRNA som sedan amplifierades med PCR för att skapa en insättning för ligering. För sin upptäckt av enzymet tilldelades Howard Temin och David Baltimore 1975 Nobelpriset i medicin och fysiologi, som de delade med Renato Dulbecco.
Kloning av PCR-produkter. Tillkomsten av PCR innebar att forskare nu kunde klona gener och DNA-segment med begränsad kunskap om amplikonsekvensen. Det rådde dock ingen större enighet om den optimala metoden för PCR-produktberedning för effektiv ligering till kloningsvektorer.
Flera olika metoder användes ursprungligen för kloning av PCR-produkter. Den enklaste och fortfarande vanligaste metoden för kloning av PCR-produkter är att införa restriktionsställen i PCR-produktens ändar (31). Detta möjliggör direkt, riktad kloning av insatsen i vektorn efter restriktionssmältning. Kloning med trubbiga ändar utvecklades för att direkt ligera PCR-produkter som genererats av polymeraser som ger trubbiga ändar, eller insättningar som konstruerats för att ha restriktionsställen som lämnar trubbiga ändar när insättningen smälts. Detta var användbart vid kloning av DNA-fragment som inte innehöll restriktionsställen som var kompatibla med vektorn (32).
Kort efter införandet av PCR introducerades PCR med överlappande förlängning (overlap extension PCR) som en metod för att sammanfoga PCR-produkter till en sammanhängande DNA-sekvens (33). I denna metod amplifieras DNA-insatsen genom PCR med hjälp av primers som genererar en PCR-produkt som innehåller överlappande områden med vektorn. Vektorn och insatsen blandas sedan, denatureras och annealas, vilket möjliggör hybridisering av insatsen till vektorn. En andra PCR-omgång genererar rekombinanta DNA-molekyler av vektorn som innehåller insatsen. PCR med överlappningsutvidgning gjorde det möjligt för forskare att sätta ihop stora gener som inte enkelt kunde amplifieras med traditionella PCR-metoder. Overlap extension PCR användes också för att införa mutationer i gensekvenser (34).
Figur 2. Översikt över PCR

Utveckling av specialiserade kloningstekniker.
I ett försök att ytterligare förbättra effektiviteten av molekylär kloning utvecklades flera specialiserade verktyg och tekniker som utnyttjade egenskaperna hos unika enzymer.
TA-kloning. En metod utnyttjade en egenskap hos Taq DNA-polymeras, det första värmestabila polymeraset som användes för PCR. Under amplifieringen lägger Taq till en enda 3´ dA-nukleotid i slutet av varje PCR-produkt. PCR-produkten kan lätt ligeras in i en vektor som har skurits och konstruerats så att den innehåller enstaka T-rester på varje sträng. Flera företag har marknadsfört tekniken och säljer kit som innehåller kloningsvektorer som redan är linjäriserade och ”svansade”.
LIC. Ligationsoberoende kloning (LIC), som namnet antyder, gör det möjligt att sammanfoga DNA-molekyler i avsaknad av DNA-ligas. LIC utförs vanligen med T4 DNA-polymeras, som används för att generera enkelsträngade DNA-överhäng, >12 nukleotider långa, på både det linjäriserade vektor-DNA:t och inslaget som ska klonas (35). När de blandas ihop annealiserar vektorn och insatsen genom den långa sträckan av kompatibla ändar. Längden på de kompatibla ändarna är tillräcklig för att hålla ihop molekylen i avsaknad av ligas, även under transformationen. När de väl har transformerats repareras luckorna in vivo. Det finns flera olika kommersiellt tillgängliga produkter för LIC.USER-kloning.
USER-kloning utvecklades först i början av 1990-talet som en restriktionsenzym- och ligasoberoende kloningsmetod (36). När metoden först utformades byggde den på att man använde PCR-primers som innehöll en ~12 nukleotisk 5´ svans, där minst fyra desoxythymidinbaser hade bytts ut mot desoxyuridiner. PCR-produkten behandlades med uracil-DNA-glykosidas (UDG) och endonukleas VIII, vilket avlägsnar uracilbaserna och lämnar en 3´överlappning som kan annealiseras till en likadant behandlad vektor. NEB säljer enzymet USER för ligas- och restriktionsenzymoberoende kloningsreaktioner.
Framtida trender
Molekylär kloning har utvecklats från kloning av ett enda DNA-fragment till sammansättning av flera DNA-komponenter i en enda sammanhängande DNA-sträcka. Ny och framväxande teknik syftar till att omvandla kloning till en process som är lika enkel som att arrangera ”block” av DNA bredvid varandra.
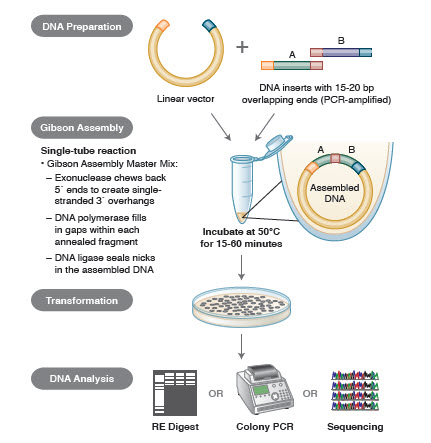
DNA-monteringsmetoder. Många nya, eleganta tekniker gör det möjligt att sammanfoga flera DNA-fragment i en enrörsreaktion. Fördelarna med dessa tekniker är att de är standardiserade, sömlösa och mestadels sekvensoberoende. Dessutom gör möjligheten att samla flera DNA-fragment i ett rör en serie tidigare oberoende restriktions-/ligeringsreaktioner till ett strömlinjeformat, effektivt förfarande.
Differenta tekniker och produkter för genmontering inkluderar SLIC (Sequence and Ligase Independent Cloning), Gibson Assembly (NEB), GeneArt® Seamless Cloning (Life Technologies) och Gateway® Cloning (Invitrogen) (35,37,38).
I DNA-montering amplifieras block av DNA som skall monteras med PCR. Därefter konstrueras de DNA-fragment som ska monteras intill varandra så att de innehåller block av komplementära sekvenser som ska ligeras ihop. Dessa kan vara kompatibla kohesiva ändar, som de som används för Gibson Assembly, eller regioner som innehåller igenkänningsställen för platsspecifika rekombinaser (Gateway). Det enzym som används för DNA-ligering kommer att känna igen och sammanfoga varje uppsättning kompatibla regioner och skapa en enda sammanhängande DNA-molekyl i en reaktion.
Figur 3. Översikt över kloningsmetoden Gibson Assembly kloning
Syntetisk biologi. DNA-syntes är ett område inom syntetisk biologi som för närvarande revolutionerar rekombinant DNA-teknik. Även om en komplett gen först syntetiserades in vitro 1972 (40), blev DNA-syntes av stora DNA-molekyler inte verklighet förrän i början av 2000-talet, då forskare började syntetisera hela genomer in vitro (41,42). Dessa tidiga experiment tog flera år att genomföra, men tekniken påskyndar möjligheten att syntetisera stora DNA-molekyler.
Slutsats
Under de senaste 40 åren har molekylär kloning utvecklats från att mödosamt isolera och sätta ihop två DNA-bitar, följt av intensiv screening av potentiella kloner, till att sömlöst sätta ihop upp till 10 DNA-fragment med anmärkningsvärd effektivitet på bara några timmar, eller att designa DNA-molekyler in silico och syntetisera dem in vitro. Tillsammans ger all denna teknik molekylärbiologerna en häpnadsväckande kraftfull verktygslåda för att utforska, manipulera och utnyttja DNA, vilket kommer att vidga vetenskapens horisonter ytterligare. Bland möjligheterna finns utveckling av säkrare rekombinanta proteiner för behandling av sjukdomar, förbättring av genterapi (43) och snabbare produktion, validering och lansering av nya vacciner (44). Men i slutändan begränsas potentialen endast av vår fantasi.
Rebecca Tirabassi är biträdande redaktör på Bitesizebio.com.
Se vår tekniska guide för molekylär kloning
.