La clonazione molecolare, un termine che è venuto a significare la creazione di molecole di DNA ricombinante, ha stimolato il progresso in tutte le scienze della vita. A partire dagli anni ’70, con la scoperta delle endonucleasi di restrizione – enzimi che tagliano selettivamente e specificamente le molecole di DNA – la tecnologia del DNA ricombinante ha visto una crescita esponenziale sia nell’applicazione che nella sofisticazione, producendo strumenti sempre più potenti per la manipolazione del DNA. La clonazione dei geni è ora così semplice ed efficiente che è diventata una tecnica di laboratorio standard. Questo ha portato ad un’esplosione nella comprensione della funzione dei geni negli ultimi decenni. Le tecnologie emergenti promettono possibilità ancora maggiori, come permettere ai ricercatori di cucire insieme senza soluzione di continuità più frammenti di DNA e trasformare i plasmidi risultanti in batteri, in meno di due ore, o l’uso di cassette di geni scambiabili, che possono essere facilmente spostati tra diversi costrutti, per massimizzare la velocità e la flessibilità. Nel prossimo futuro, la clonazione molecolare vedrà probabilmente l’emergere di un nuovo paradigma, con tecniche di biologia sintetica che permetteranno la sintesi chimica in vitro di qualsiasi costrutto di DNA specificato in silico. Questi progressi dovrebbero consentire una più rapida costruzione e iterazione dei cloni di DNA, accelerando lo sviluppo di vettori di terapia genica, processi di produzione di proteine ricombinanti e nuovi vaccini.
Rebecca Tirabassi, Bitesize Bio.
- Introduzione
- Storia della clonazione
- La base della clonazione molecolare
- Figura 1. Flusso di lavoro tradizionale del clonaggio
- Building on the Groundwork
- Figura 2. Panoramica della PCR
- Tendenze future
- Figura 3. Panoramica del metodo di clonazione Gibson Assembly
- Conclusione
- Vedi la nostra Guida tecnica alla clonazione molecolare
Introduzione
La clonazione molecolare si riferisce all’isolamento di una sequenza di DNA da qualsiasi specie (spesso un gene), e il suo inserimento in un vettore per la propagazione, senza alterazione della sequenza originale del DNA. Una volta isolati, i cloni molecolari possono essere utilizzati per generare molte copie del DNA per l’analisi della sequenza genica, e/o per esprimere la proteina risultante per lo studio o l’utilizzo della funzione della proteina. I cloni possono anche essere manipolati e mutati in vitro per alterare l’espressione e la funzione della proteina.
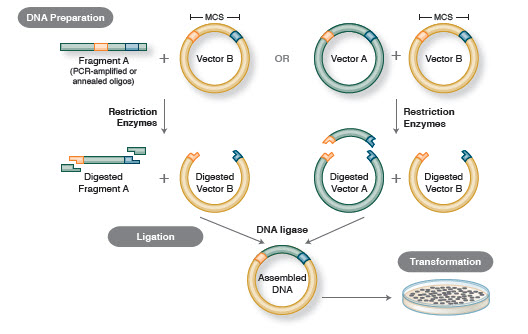
Il flusso di lavoro di base della clonazione include quattro fasi:
- Isolamento di frammenti di DNA target (spesso indicati come inserti)
- Ligazione degli inserti in un vettore di clonazione appropriato, creando molecole ricombinanti (es, plasmidi)
- Trasformazione di plasmidi ricombinanti in batteri o altri ospiti adatti per la propagazione
- Selezione di ospiti contenenti il plasmide ricombinante desiderato
Questi quattro passi rivoluzionari sono stati accuratamente messi insieme ed eseguiti da più laboratori, a partire dalla fine degli anni ’60 e l’inizio degli anni ’70. Un riassunto delle scoperte che comprendono la clonazione molecolare tradizionale è descritto nelle pagine seguenti.
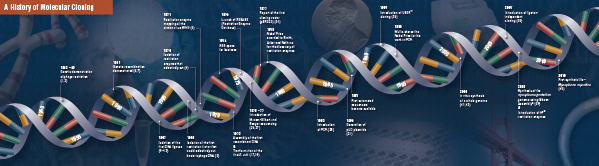
Storia della clonazione
La base della clonazione molecolare
Taglio (digestione). La tecnologia del DNA ricombinante è emersa per la prima volta alla fine degli anni ’60, con la scoperta di enzimi che potevano tagliare e unire specificamente le molecole di DNA a doppio filamento. Infatti, già nel 1952, due gruppi hanno osservato indipendentemente che i batteri codificavano un “fattore di restrizione” che impediva ai batteriofagi di crescere in certi ospiti (1,2). Ma la natura del fattore non fu scoperta fino al 1968, quando Arber e Linn riuscirono a isolare un enzima, chiamato fattore di restrizione, che tagliava selettivamente il DNA esogeno, ma non quello batterico (3). Questi studi identificarono anche un enzima metilasi che proteggeva il DNA batterico dagli enzimi di restrizione.
Poco dopo la scoperta di Arber e Linn, Smith estese e confermò questi studi isolando un enzima di restrizione da Haemophilus influenza. Egli dimostrò che l’enzima tagliava selettivamente il DNA nel mezzo di uno specifico tratto di 6 paia di basi del DNA; una caratteristica di certi enzimi di restrizione è la loro propensione a tagliare il substrato del DNA in o vicino a specifiche, spesso palindromiche, sequenze di “riconoscimento” (4).
Il pieno potere degli enzimi di restrizione non è stato realizzato fino a quando gli enzimi di restrizione e l’elettroforesi del gel non sono stati utilizzati per mappare il genoma del Simian Virus 40 (SV40) (5). Per queste scoperte seminali, Werner Arber, Hamilton Smith e Daniel Nathans hanno condiviso il premio Nobel per la medicina del 1978.
Figura 1. Flusso di lavoro tradizionale del clonaggio
Assembling (Ligation). Molto simile alla scoperta degli enzimi che tagliano il DNA, la scoperta di un enzima che potrebbe unire il DNA è stata preceduta da osservazioni precedenti e salienti. All’inizio degli anni ’60, due gruppi scoprirono che la ricombinazione genetica poteva avvenire tramite la rottura e la legatura di molecole di DNA (6,7), seguita da vicino dall’osservazione che il DNA lineare dei batteriofagi viene rapidamente convertito in cerchi covalentemente chiusi dopo l’infezione dell’ospite (8). Solo due anni dopo, cinque gruppi hanno isolato indipendentemente le DNA ligasi e dimostrato la loro capacità di assemblare due pezzi di DNA (9-13).
Non molto tempo dopo la scoperta degli enzimi di restrizione e delle DNA ligasi, fu realizzata la prima molecola di DNA ricombinante. Nel 1972, Berg tagliò e legò separatamente un pezzo di DNA di batteriofago lambda o l’operone del galattosio di E. coli con il DNA SV40 per creare le prime molecole di DNA ricombinante (14). Questi studi aprirono la strada al concetto che, a causa della natura universale del DNA, il DNA di qualsiasi specie poteva essere unito. Nel 1980, Paul Berg ha condiviso il premio Nobel per la chimica con Walter Gilbert e Frederick Sanger (gli sviluppatori del sequenziamento del DNA), per “i suoi studi fondamentali sulla biochimica degli acidi nucleici, con particolare riguardo al DNA ricombinante”.
Trasformazione. La tecnologia del DNA ricombinante sarebbe gravemente limitata, e la clonazione molecolare impossibile, senza i mezzi per propagare e isolare la molecola di DNA appena costruita. La capacità di trasformare i batteri, o di indurre l’assorbimento, l’incorporazione e l’espressione di materiale genetico estraneo, fu dimostrata per la prima volta da Griffith quando trasformò un ceppo non letale di batteri in un ceppo letale mescolando il ceppo non letale con batteri letali inattivati dal calore (15). Tuttavia, la natura del “principio di trasformazione” che trasmetteva la letalità non fu compresa fino al 1944. Nello stesso anno, Avery, Macleod e McCarty dimostrarono che il DNA, e non la proteina, era responsabile dell’induzione del fenotipo letale (16).
Inizialmente, si credeva che il comune ceppo batterico da laboratorio, E. coli, fosse refrattario alla trasformazione, finché Mandel e Higa dimostrarono che il trattamento di E. coli con cloruro di calcio induceva l’assorbimento del DNA batteriofago (17). Cohen ha applicato questo principio, nel 1972, quando ha aperto la strada alla trasformazione di batteri con plasmidi per conferire resistenza antibiotica ai batteri (18).
L’esperimento definitivo: digestione, legatura e trasformazione di una molecola di DNA ricombinante fu eseguito da Boyer, Cohen e Chang nel 1973, quando digerirono il plasmide pSC101 con EcoRI, legarono il frammento linearizzato ad un altro plasmide limitato dall’enzima e trasformarono la molecola ricombinante risultante in E. coli, conferendo tetraciclina. coli, conferendo ai batteri la resistenza alla tetraciclina (19), gettando così le basi per la maggior parte del lavoro sul DNA ricombinante da allora.
Building on the Groundwork
Mentre gli scienziati avevano scoperto e applicato tutti i principi di base per creare e propagare il DNA ricombinante nei batteri, il processo era inefficiente. Le preparazioni degli enzimi di restrizione erano inaffidabili a causa di procedure di purificazione non standardizzate, i plasmidi per la clonazione erano ingombranti, difficili da lavorare e in numero limitato, e gli esperimenti erano limitati dalla quantità di DNA inserito che poteva essere isolato. La ricerca nei decenni successivi portò a miglioramenti nelle tecniche e negli strumenti disponibili per la clonazione molecolare.
Prima progettazione dei vettori.
Sviluppo del primo vettore standardizzato. Gli scienziati che lavoravano nel laboratorio di Boyer hanno riconosciuto la necessità di un plasmide di clonazione generale, un plasmide compatto con siti di restrizione unici per la clonazione in DNA estraneo e l’espressione di geni di resistenza agli antibiotici per la selezione dei batteri trasformati. Nel 1977, hanno descritto il primo vettore progettato per scopi di clonazione, pBR322 (20). Questo vettore era piccolo, ~4 kilobasi di dimensione, e aveva due geni di resistenza agli antibiotici per la selezione.
Vettori con selezione a bordo e rese più elevate. Anche se la selezione antibiotica impediva ai batteri non trasformati di crescere, i plasmidi che si rilegavano senza frammenti di DNA dell’inserto (auto-legatura) potevano ancora conferire resistenza antibiotica ai batteri. Pertanto, trovare i cloni batterici corretti contenenti la molecola di DNA ricombinante desiderata potrebbe richiedere molto tempo.
Vieira e Messing hanno ideato uno strumento di screening per identificare le colonie batteriche contenenti plasmidi con inserti di DNA. Basandosi sul plasmide pBR322, crearono la serie di plasmidi pUC, che conteneva un sistema di “screening blu/bianco” (21). Il posizionamento di un sito di clonazione multiplo (MCS) contenente diversi siti di restrizione unici all’interno del gene LacZ´ ha permesso ai ricercatori di cercare colonie batteriche contenenti plasmidi con l’inserto di DNA straniero. Quando i batteri venivano piastrati sui supporti corretti, le colonie bianche contenevano plasmidi con inserti, mentre le colonie blu contenevano plasmidi senza inserti. I plasmidi pUC avevano un ulteriore vantaggio rispetto ai vettori esistenti: contenevano una mutazione che determinava un numero di copie più elevato, aumentando così la resa dei plasmidi.
Miglioramento dei digest di restrizione. I primi lavori con gli enzimi di restrizione furono ostacolati dalla purezza della preparazione dell’enzima e dalla mancanza di comprensione dei requisiti del buffer per ogni enzima. Nel 1975, New England Biolabs (NEB) divenne la prima azienda a commercializzare enzimi di restrizione prodotti da una fonte ricombinante. Questo ha permesso di ottenere rendimenti più elevati, una maggiore purezza, coerenza da lotto a lotto e prezzi più bassi. Attualmente, oltre 4.000 enzimi di restrizione, che riconoscono oltre 300 sequenze diverse, sono stati scoperti dagli scienziati di tutto il mondo. NEB fornisce attualmente oltre 230 di queste specificità.
NEB è stata anche una delle prime aziende a sviluppare un sistema a quattro tamponi standardizzato e a caratterizzare tutte le sue attività enzimatiche in questo sistema di tamponi. Questo ha portato a una migliore comprensione di come condurre un doppio digest, o la digestione del DNA con due enzimi contemporaneamente. La ricerca successiva ha portato allo sviluppo di sistemi one-buffer, che sono compatibili con gli enzimi di restrizione più comuni (come il CutSmart™ Buffer di NEB).
Con l’avvento di librerie di enzimi di restrizione disponibili in commercio con specificità di sequenza note, gli enzimi di restrizione sono diventati un potente strumento per lo screening di potenziali cloni di DNA ricombinante. Il “digest diagnostico” era, ed è ancora, una delle tecniche più comuni usate nel clonaggio molecolare.
Preparazione di vettori e inserti. L’efficienza e la versatilità del clonaggio sono state migliorate anche dallo sviluppo di diverse tecniche per la preparazione dei vettori prima della legatura. Furono isolate le fosfatasi alcaline che potevano rimuovere i gruppi fosfato 3′ e 5′ dalle estremità del DNA. Fu presto scoperto che il trattamento dei vettori con la fosfatasi di vitello intestinale (CIP) de fosforilava le estremità del DNA e impediva l’auto-legatura del vettore, aumentando il recupero dei plasmidi con l’inserto (24).
L’enzima CIP si dimostrò difficile da inattivare, e qualsiasi attività residua portava alla de fosforilazione del DNA inserito e all’inibizione della reazione di legatura. La scoperta delle fosfatasi alcaline termolabili, come la fosfatasi alcalina ricombinante di gamberetti (rSAP) e la fosfatasi antartica (AP) (entrambe vendute da NEB), diminuì i passaggi e il tempo necessario, poiché un semplice spostamento di temperatura inattiva l’enzima prima della fase di legatura (25).
Arriva il sequenziamento del DNA. Il sequenziamento del DNA è stato sviluppato alla fine degli anni ’70, quando furono ideati due metodi concorrenti. Maxam e Gilbert svilupparono il “metodo di sequenziamento chimico”, che si basava sulla modifica chimica del DNA e la successiva scissione in basi specifiche (26). Allo stesso tempo, Sanger e colleghi pubblicarono il “metodo della terminazione a catena”, che divenne il metodo usato dalla maggior parte dei ricercatori (27). Il metodo Sanger divenne rapidamente automatizzato, e i primi sequenziatori automatici furono venduti nel 1987.
La capacità di determinare la sequenza di un tratto di DNA aumentò l’affidabilità e la versatilità della clonazione molecolare. Una volta clonati, gli scienziati potevano sequenziare i cloni per identificare definitivamente la molecola ricombinante corretta, identificare nuovi geni o mutazioni nei geni, e progettare facilmente oligonucleotidi basati sulla sequenza nota per ulteriori esperimenti.
L’impatto della reazione a catena della polimerasi. Uno dei problemi della clonazione molecolare nei primi anni era quello di ottenere abbastanza DNA da inserire nel vettore. Nel 1983, Mullis ideò una tecnica che risolse questo problema e rivoluzionò la clonazione molecolare (28). Ha amplificato un tratto di DNA bersaglio usando primer opposti per amplificare entrambi i filamenti complementari di DNA, simultaneamente. Attraverso cicli di denaturazione, ricottura e polimerizzazione, dimostrò di poter amplificare esponenzialmente una singola copia di DNA. La reazione a catena della polimerasi, o PCR, rese possibile amplificare e clonare i geni da quantità di DNA precedentemente inadeguate. Per questa scoperta, Kary Mullis ha condiviso il premio Nobel 1993 per la chimica “per i contributi allo sviluppo di metodi nell’ambito della chimica basata sul DNA”.
Nel 1970, Temin e Baltimore scoprirono indipendentemente la trascrittasi inversa nei virus, un enzima che converte l’RNA in DNA (29,30). Poco dopo lo sviluppo della PCR, la trascrizione inversa è stata accoppiata alla PCR (RT-PCR) per permettere la clonazione dell’RNA messaggero (mRNA). La trascrizione inversa veniva usata per creare una copia di DNA (cDNA) dell’mRNA che veniva successivamente amplificato dalla PCR per creare un inserto per la legatura. Per la loro scoperta dell’enzima, Howard Temin e David Baltimore hanno ricevuto il premio Nobel per la medicina e la fisiologia nel 1975, che hanno condiviso con Renato Dulbecco.
Clonazione di prodotti PCR. L’avvento della PCR significava che i ricercatori potevano ora clonare geni e segmenti di DNA con una conoscenza limitata della sequenza degli ampliconi. Tuttavia, c’era poco consenso sul metodo ottimale di preparazione dei prodotti di PCR per una legatura efficiente nei vettori di clonazione.
Diversi metodi sono stati inizialmente utilizzati per clonare i prodotti PCR. Il metodo più semplice, e ancora il più comune, per clonare i prodotti PCR è attraverso l’introduzione di siti di restrizione sulle estremità del prodotto PCR (31). Questo permette la clonazione diretta e direzionale dell’inserto nel vettore dopo la digestione di restrizione. La clonazione Blunt-ended è stata sviluppata per legare direttamente i prodotti PCR generati da polimerasi che producevano estremità smussate, o inserti ingegnerizzati per avere siti di restrizione che lasciavano estremità smussate una volta che l’inserto veniva digerito. Questo era utile per clonare frammenti di DNA che non contenevano siti di restrizione compatibili con il vettore (32).
Poco dopo l’introduzione della PCR, fu introdotta la PCR con estensione a sovrapposizione come metodo per assemblare i prodotti della PCR in una sequenza di DNA contigua (33). In questo metodo, l’inserto di DNA viene amplificato tramite PCR utilizzando primer che generano un prodotto PCR contenente regioni sovrapposte al vettore. Il vettore e l’inserto vengono poi mescolati, denaturati e anneificati, permettendo l’ibridazione dell’inserto al vettore. Un secondo ciclo di PCR genera molecole di DNA ricombinante del vettore contenente l’inserto. La PCR con estensione a sovrapposizione ha permesso ai ricercatori di mettere insieme grandi geni che non potevano essere facilmente amplificati con i metodi tradizionali di PCR. La PCR di estensione della sovrapposizione è stata anche usata per introdurre mutazioni nelle sequenze di geni (34).
Figura 2. Panoramica della PCR

Sviluppo di tecniche di clonazione specializzate.
Nello sforzo di migliorare ulteriormente l’efficienza della clonazione molecolare, furono sviluppati diversi strumenti e tecniche specializzati che sfruttavano le proprietà di enzimi unici.
TA Cloning. Un approccio ha sfruttato una proprietà della Taq DNA Polymerase, la prima polimerasi stabile al calore usata per la PCR. Durante l’amplificazione, la Taq aggiunge un singolo nucleotide 3´ dA alla fine di ogni prodotto PCR. Il prodotto di PCR può essere facilmente legato in un vettore che è stato tagliato e ingegnerizzato per contenere singoli residui T su ogni filamento. Diverse aziende hanno commercializzato questa tecnica e vendono kit contenenti vettori di clonazione che sono già linearizzati e “codificati”.
LIC. Ligation independent cloning (LIC), come dice il nome, permette l’unione di molecole di DNA in assenza di DNA ligasi. La LIC viene comunemente eseguita con la DNA polimerasi T4, che viene utilizzata per generare delle sporgenze di DNA a singolo filamento, lunghe >12 nucleotidi, sia sul DNA vettore linearizzato che sull’inserto da clonare (35). Quando vengono mescolati insieme, il vettore e l’inserto si annebbiano attraverso il lungo tratto di estremità compatibili. La lunghezza delle estremità compatibili è sufficiente a tenere insieme la molecola in assenza di ligasi, anche durante la trasformazione. Una volta trasformata, le lacune vengono riparate in vivo. Ci sono diversi prodotti disponibili in commercio per la clonazione LIC.USER.
La clonazione USER è stata sviluppata nei primi anni ’90 come metodo di clonazione indipendente dagli enzimi di restrizione e dalla ligasi (36). Quando fu concepito per la prima volta, il metodo si basava sull’uso di primer PCR che contenevano una coda di ~12 nucleotidi 5´, in cui almeno quattro basi di deossitimidina erano state sostituite con deossiuridine. Il prodotto di PCR è stato trattato con uracile DNA glicosidasi (UDG) e Endonucleasi VIII, che elimina le basi di uracile e lascia una sovrapposizione 3 ‘che può essere annealed a un vettore trattato in modo simile. NEB vende l’enzima USER per reazioni di clonazione indipendenti da ligasi ed enzimi di restrizione.
Tendenze future
La clonazione molecolare è progredita dalla clonazione di un singolo frammento di DNA all’assemblaggio di più componenti di DNA in un singolo tratto contiguo di DNA. Le nuove tecnologie emergenti cercano di trasformare la clonazione in un processo semplice come la disposizione di “blocchi” di DNA uno accanto all’altro.
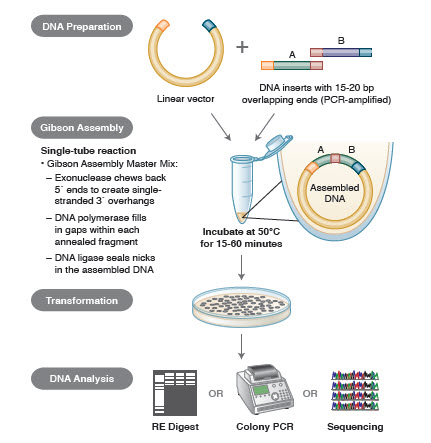
Metodi di assemblaggio del DNA. Molte nuove ed eleganti tecnologie permettono l’assemblaggio di frammenti multipli di DNA in una reazione con un solo tubo. I vantaggi di queste tecnologie sono che sono standardizzate, senza soluzione di continuità e per lo più indipendenti dalla sequenza. Inoltre, la capacità di assemblare più frammenti di DNA in un tubo trasforma una serie di reazioni di restrizione/ligazione precedentemente indipendenti in una procedura snella ed efficiente.
Diverse tecniche e prodotti per l’assemblaggio genico includono SLIC (Sequence and Ligase Independent Cloning), Gibson Assembly (NEB), GeneArt® Seamless Cloning (Life Technologies) e Gateway® Cloning (Invitrogen) (35,37,38).
Nell’assemblaggio del DNA, i blocchi di DNA da assemblare sono amplificati tramite PCR. Poi, i frammenti di DNA da assemblare adiacenti l’uno all’altro sono ingegnerizzati per contenere blocchi di sequenze complementari che saranno legati insieme. Queste potrebbero essere estremità coesive compatibili, come quelle usate per il Gibson Assembly, o regioni contenenti siti di riconoscimento per ricombinasi sito-specifiche (Gateway). L’enzima usato per la legatura del DNA riconoscerà e assemblerà ogni serie di regioni compatibili, creando una singola molecola di DNA contigua in una sola reazione.
Figura 3. Panoramica del metodo di clonazione Gibson Assembly
Biologia sintetica. La sintesi del DNA è un’area della biologia sintetica che sta attualmente rivoluzionando la tecnologia del DNA ricombinante. Anche se un gene completo è stato sintetizzato per la prima volta in vitro nel 1972 (40), la sintesi del DNA di grandi molecole di DNA non è diventata una realtà fino ai primi anni 2000, quando i ricercatori hanno iniziato a sintetizzare interi genomi in vitro (41,42). Questi primi esperimenti hanno richiesto anni per essere completati, ma la tecnologia sta accelerando la capacità di sintetizzare grandi molecole di DNA.
Conclusione
Negli ultimi 40 anni, la clonazione molecolare è progredita dall’arduo isolamento e assemblaggio di due pezzi di DNA, seguito da un intenso screening di potenziali cloni, all’assemblaggio senza soluzione di continuità fino a 10 frammenti di DNA con notevole efficienza in poche ore, o alla progettazione di molecole di DNA in silico e alla loro sintesi in vitro. Insieme, tutte queste tecnologie danno ai biologi molecolari una cassetta degli attrezzi sorprendentemente potente per esplorare, manipolare e sfruttare il DNA, che amplierà ulteriormente gli orizzonti della scienza. Tra le possibilità ci sono lo sviluppo di proteine ricombinanti più sicure per il trattamento delle malattie, il miglioramento della terapia genica (43), e una più rapida produzione, validazione e rilascio di nuovi vaccini (44). Ma in definitiva, il potenziale è limitato solo dalla nostra immaginazione.
Rebecca Tirabassi è un Assistant Editor di Bitesizebio.com.