Molekulární klonování, termín, který označuje vytváření rekombinantních molekul DNA, podnítilo pokrok v celé oblasti věd o živé přírodě. Počínaje 70. lety 20. století, kdy byly objeveny restrikční endonukleázy – enzymy, které selektivně a specificky stříhají molekuly DNA – zaznamenala technologie rekombinantní DNA exponenciální růst jak v oblasti použití, tak v oblasti sofistikovanosti, což přináší stále výkonnější nástroje pro manipulaci s DNA. Klonování genů je nyní tak jednoduché a účinné, že se stalo standardní laboratorní technikou. To vedlo v posledních desetiletích k prudkému rozvoji poznání funkce genů. Nově vznikající technologie slibují ještě větší možnosti, například umožňují výzkumným pracovníkům bezproblémově spojit dohromady několik fragmentů DNA a výsledné plazmidy transformovat do bakterií za méně než dvě hodiny nebo používat vyměnitelné genové kazety, které lze snadno přesouvat mezi různými konstrukcemi, aby se maximalizovala rychlost a flexibilita. V blízké budoucnosti se v oblasti molekulárního klonování pravděpodobně objeví nové paradigma s technikami syntetické biologie, které umožní chemickou syntézu in vitro jakéhokoli in silico specifikovaného konstruktu DNA. Tyto pokroky by měly umožnit rychlejší konstrukci a iteraci klonů DNA, což urychlí vývoj vektorů pro genovou terapii, postupy výroby rekombinantních proteinů a nových vakcín.
Rebecca Tirabassi, Bitesize Bio.
Úvod
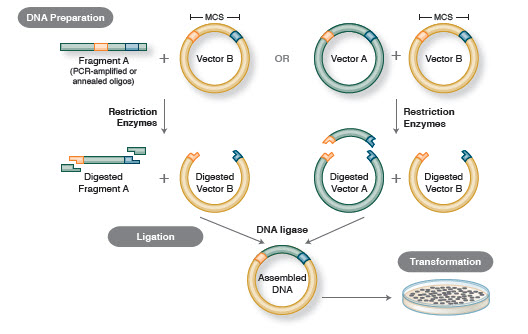
Molekulárním klonováním se rozumí izolace sekvence DNA z jakéhokoli druhu (často genu) a její vložení do vektoru za účelem množení, aniž by došlo ke změně původní sekvence DNA. Po izolaci lze molekulární klony použít k vytvoření mnoha kopií DNA pro analýzu sekvence genu a/nebo k expresi výsledného proteinu pro studium nebo využití funkce proteinu. S klony lze také manipulovat a mutovat je in vitro za účelem změny exprese a funkce proteinu.
Základní pracovní postup klonování zahrnuje čtyři kroky:
- Izolace cílových fragmentů DNA (často označovaných jako inserty)
- Ligace insertů do vhodného klonovacího vektoru, čímž se vytvoří rekombinantní molekuly (např, plazmidy)
- Transformace rekombinantních plazmidů do bakterií nebo jiného vhodného hostitele pro množení
- Screening/výběr hostitelů obsahujících zamýšlený rekombinantní plazmid
Tyto čtyři průlomové kroky byly pečlivě poskládány dohromady a prováděny mnoha laboratořemi, počínaje koncem 60. a začátkem 70. let 20. století. Shrnutí objevů, které tvoří tradiční molekulární klonování, je popsáno na následujících stránkách.
Historie klonování
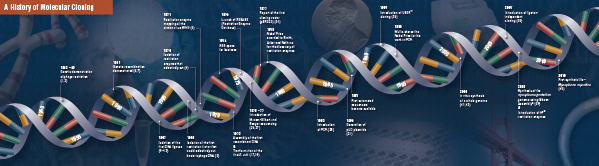
Základy molekulárního klonování
Stříhání (digesce). Technologie rekombinantní DNA se poprvé objevila koncem 60. let 20. století, kdy byly objeveny enzymy, které dokázaly specificky řezat a spojovat dvouřetězcové molekuly DNA. Ve skutečnosti již v roce 1952 dvě skupiny nezávisle na sobě pozorovaly, že bakterie kódují „restrikční faktor“, který zabraňuje růstu bakteriofágů v určitých hostitelích (1,2). Povaha tohoto faktoru však byla objevena až v roce 1968, kdy se Arberovi a Linnovi podařilo izolovat enzym, označovaný jako restrikční faktor, který selektivně stříhal exogenní DNA, ale ne bakteriální DNA (3). Tyto studie také identifikovaly enzym metylázu, který chránil bakteriální DNA před restrikčními enzymy.
Krátce po objevu Arbera a Linna Smith rozšířil a potvrdil tyto studie izolací restrikčního enzymu z Haemophilus influenza. Prokázal, že tento enzym selektivně stříhá DNA uprostřed specifického úseku DNA o délce 6 párů bází; jednou z charakteristik některých restrikčních enzymů je jejich sklon stříhat substrát DNA ve specifických, často palindromických „rozpoznávacích“ sekvencích nebo v jejich blízkosti (4).
Plnou sílu restrikčních enzymů jsme si uvědomili až poté, co byly restrikční enzymy a gelová elektroforéza použity k mapování genomu viru Simian Virus 40 (SV40) (5). Za tyto zásadní objevy se Werner Arber, Hamilton Smith a Daniel Nathans v roce 1978 podělili o Nobelovu cenu za medicínu.
Obrázek 1. Tradiční pracovní postup klonování
Assembling (Ligation). Podobně jako objevu enzymů, které stříhají DNA, předcházelo objevu enzymu, který dokáže spojovat DNA, dřívější významné pozorování. Na počátku 60. let 20. století dvě skupiny objevily, že ke genetické rekombinaci může docházet prostřednictvím přerušení a vazby molekul DNA (6,7), na což těsně navázalo pozorování, že lineární bakteriofágová DNA se po infekci hostitele rychle mění na kovalentně uzavřené kruhy (8). Jen o dva roky později pět skupin nezávisle na sobě izolovalo DNA ligázy a prokázalo jejich schopnost sestavovat dva kusy DNA (9-13).
Nedlouho po objevu restrikčních enzymů a ligáz DNA byla vytvořena první rekombinantní molekula DNA. V roce 1972 Berg odděleně rozstříhal a podvázal kus DNA bakteriofága lambda nebo galaktózového operonu E. coli s DNA SV40 a vytvořil první rekombinantní molekuly DNA (14). Tyto studie byly průkopníkem koncepce, že vzhledem k univerzální povaze DNA lze spojit DNA jakéhokoli druhu. V roce 1980 se Paul Berg podělil o Nobelovu cenu za chemii s Walterem Gilbertem a Frederickem Sangerem (tvůrci sekvenování DNA) za „základní studie biochemie nukleových kyselin se zvláštním zřetelem na rekombinantní DNA“.
Transformace. Technologie rekombinantní DNA by byla značně omezená a molekulární klonování nemožné bez prostředků k rozmnožování a izolaci nově vytvořené molekuly DNA. Schopnost transformovat bakterie neboli vyvolat příjem, začlenění a expresi cizího genetického materiálu poprvé prokázal Griffith, když transformoval neletální kmen bakterií na letální kmen smícháním neletálního kmene s tepelně inaktivovanými letálními bakteriemi (15). Povaha „transformačního principu“, který přenáší letalitu, však byla pochopena až v roce 1944. V témže roce Avery, Macleod a McCarty prokázali, že za vyvolání letálního fenotypu je zodpovědná DNA, a nikoliv bílkovina (16).
Zpočátku se mělo za to, že běžný laboratorní bakteriální kmen E. coli je vůči transformaci odolný, dokud Mandel a Higa neprokázali, že ošetření E. coli chloridem vápenatým vyvolalo přijetí DNA bakteriofága (17). Tento princip použil Cohen v roce 1972, když jako první provedl transformaci bakterií pomocí plazmidů, aby bakteriím propůjčil odolnost vůči antibiotikům (18).
Konečný experiment: digesci, ligaci a transformaci rekombinantní molekuly DNA provedli Boyer, Cohen a Chang v roce 1973, kdy digesci plazmidu pSC101 provedli pomocí EcoRI, linearizovaný fragment podvázali na jiný plazmid s omezeným množstvím enzymů a výslednou rekombinantní molekulu transformovali do E. coli, čímž bakteriím propůjčili rezistenci vůči tetracyklinu (19), a položili tak základ většiny prací s rekombinantní DNA od té doby.
Building on the Groundwork
Přestože vědci objevili a použili všechny základní principy pro vytváření a množení rekombinantní DNA v bakteriích, byl tento proces neefektivní. Přípravky restrikčních enzymů byly nespolehlivé kvůli nestandardizovaným purifikačním postupům, plazmidy pro klonování byly těžkopádné, špatně se s nimi pracovalo a jejich počet byl omezený a experimenty byly limitovány množstvím vložené DNA, kterou bylo možné izolovat. Výzkum v průběhu několika následujících desetiletí vedl ke zlepšení technik a nástrojů dostupných pro molekulární klonování.
Raný návrh vektorů.
Vývoj prvního standardizovaného vektoru. Vědci pracující v Boyerově laboratoři rozpoznali potřebu obecného klonovacího plazmidu, kompaktního plazmidu s jedinečnými restrikčními místy pro klonování cizí DNA a expresi genů rezistence vůči antibiotikům pro selekci transformovaných bakterií. V roce 1977 popsali první vektor určený pro klonovací účely, pBR322 (20). Tento vektor byl malý, o velikosti ~4 kilobáze, a měl dva geny antibiotické rezistence pro selekci.
Vektory s palubní selekcí a vyššími výtěžky. Ačkoli selekce antibiotiky zabránila růstu netransformovaných bakterií, plazmidy, které se relizovaly bez vložených fragmentů DNA (samoligace), mohly bakteriím stále propůjčovat rezistenci vůči antibiotikům. Proto mohlo být nalezení správných bakteriálních klonů obsahujících požadovanou rekombinantní molekulu DNA časově náročné.
Vieira a Messing navrhli screeningový nástroj pro identifikaci bakteriálních kolonií obsahujících plazmidy s vloženou DNA. Na základě plazmidu pBR322 vytvořili sérii plazmidů pUC, která obsahovala „modrobílý screeningový systém“ (21). Umístění vícenásobného klonovacího místa (MCS) obsahujícího několik unikátních restrikčních míst v rámci genu LacZ´ umožnilo výzkumníkům provádět screening bakteriálních kolonií obsahujících plazmidy s cizím DNA insertem. Když byly bakterie naočkovány na správné médium, bílé kolonie obsahovaly plazmidy s inzerty, zatímco modré kolonie obsahovaly plazmidy bez inzertu. pUC plazmidy měly oproti stávajícím vektorům další výhodu; obsahovaly mutaci, která vedla k vyššímu počtu kopií, a proto zvyšovala výtěžnost plazmidů.
Zlepšení restrikčních digescí. Rané práce s restrikčními enzymy byly ztíženy čistotou enzymových preparátů a nedostatečným pochopením požadavků na pufry pro jednotlivé enzymy. V roce 1975 se společnost New England Biolabs (NEB) stala první společností, která komercializovala restrikční enzymy vyrobené z rekombinantního zdroje. To umožnilo vyšší výtěžnost, lepší čistotu, konzistenci jednotlivých šarží a nižší ceny. V současné době vědci z celého světa objevili více než 4 000 restrikčních enzymů, které rozpoznávají více než 300 různých sekvencí . Společnost NEB v současné době dodává více než 230 těchto specifických enzymů.
NEB byla také jednou z prvních společností, která vyvinula standardizovaný systém čtyř pufrů a charakterizovala všechny aktivity svých enzymů v tomto systému pufrů. To vedlo k lepšímu pochopení způsobu provádění dvojitého štěpení neboli štěpení DNA dvěma enzymy současně. Pozdější výzkum vedl k vývoji systémů s jedním pufrem, které jsou kompatibilní s nejběžnějšími restrikčními enzymy (například CutSmart™ Buffer společnosti NEB).
S příchodem komerčně dostupných knihoven restrikčních enzymů se známou sekvenční specifitou se restrikční enzymy staly mocným nástrojem pro screening potenciálních klonů rekombinantní DNA. „Diagnostické trávení“ bylo a stále je jednou z nejběžnějších technik používaných při molekulárním klonování.
Příprava vektorů a insertů. Účinnost a univerzálnost klonování se zlepšila také vývojem různých technik přípravy vektorů před ligací. Byly izolovány alkalické fosfatázy, které dokázaly odstranit 3´ a 5´ fosfátové skupiny z konců DNA . Brzy bylo zjištěno, že ošetření vektorů calf-intestinální fosfatázou (CIP) defosforyluje konce DNA a zabraňuje samoligaci vektoru, čímž se zvyšuje výtěžnost plazmidů s inzertem (24).
Enzym CIP se ukázal jako obtížně inaktivovatelný a jakákoli zbytková aktivita vedla k defosforylaci inzertní DNA a inhibici ligační reakce. Objev tepelně labilních alkalických fosfatáz, jako je rekombinantní krevetová alkalická fosfatáza (rSAP) a antarktická fosfatáza (AP) (obě prodávané společností NEB), snížil počet kroků a časovou náročnost, protože pouhý posun teploty inaktivuje enzym před krokem ligace (25).
Přichází sekvenování DNA. Sekvenování DNA bylo vyvinuto koncem 70. let 20. století, kdy byly navrženy dvě konkurenční metody. Maxam a Gilbert vyvinuli „chemickou metodu sekvenování“, která spočívala v chemické modifikaci DNA a následném štěpení na specifických bázích (26). Ve stejné době Sanger a jeho kolegové publikovali „metodu řetězového zakončení“, která se stala metodou používanou většinou výzkumníků (27). Sangerova metoda se rychle automatizovala a v roce 1987 se začaly prodávat první automatické sekvenátory.
Schopnost určit sekvenci úseku DNA zvýšila spolehlivost a univerzálnost molekulárního klonování. Po klonování mohli vědci sekvenovat klony, aby definitivně určili správnou rekombinantní molekulu, identifikovat nové geny nebo mutace v genech a snadno navrhovat oligonukleotidy na základě známé sekvence pro další experimenty.
Vliv polymerázové řetězové reakce. Jedním z problémů molekulárního klonování v prvních letech bylo získání dostatečného množství vložené DNA pro klonování do vektoru. V roce 1983 Mullis vymyslel techniku, která tento problém vyřešila a způsobila revoluci v molekulárním klonování (28). Amplifikoval úsek cílové DNA pomocí protilehlých primerů, které amplifikovaly obě komplementární vlákna DNA současně. Pomocí cyklů denaturace, žíhání a polymerace ukázal, že může exponenciálně amplifikovat jednu kopii DNA. Polymerázová řetězová reakce neboli PCR umožnila amplifikovat a klonovat geny z dříve nedostatečného množství DNA. Za tento objev se Kary Mullis v roce 1993 podělil o Nobelovu cenu za chemii „za přínos k rozvoji metod v rámci chemie založené na DNA“.
V roce 1970 Temin a Baltimore nezávisle na sobě objevili reverzní transkriptázu u virů, enzym, který převádí RNA na DNA (29,30). Krátce po vývoji PCR byla reverzní transkripce spojena s PCR (RT-PCR), což umožnilo klonování messengerové RNA (mRNA). Reverzní transkripce byla použita k vytvoření kopie DNA (cDNA) mRNA, která byla následně amplifikována pomocí PCR a vytvořila insert pro ligaci. Za objev tohoto enzymu získali Howard Temin a David Baltimore v roce 1975 Nobelovu cenu za lékařství a fyziologii, o kterou se podělili s Renatem Dulbeccem.
Klonování produktů PCR. Nástup PCR znamenal, že vědci nyní mohou klonovat geny a úseky DNA s omezenou znalostí sekvence amplikonu. Neexistovala však shoda ohledně optimální metody přípravy produktů PCR pro účinnou ligaci do klonovacích vektorů.
Původně se pro klonování produktů PCR používalo několik různých metod. Nejjednodušší a stále nejběžnější metodou klonování produktů PCR je zavedení restrikčních míst na konce produktů PCR (31). To umožňuje přímé, směrové klonování inzertu do vektoru po restrikčním štěpení. Klonování s tupými konci bylo vyvinuto pro přímou ligaci produktů PCR generovaných polymerázami, které vytvářejí tupé konce, nebo inzertů upravených tak, aby měly restrikční místa, která po natrávení inzertů zanechávají tupé konce. To bylo užitečné při klonování fragmentů DNA, které neobsahovaly restrikční místa kompatibilní s vektorem (32).
Krátce po zavedení PCR byla zavedena metoda overlap extension PCR, která umožňuje sestavit produkty PCR do jedné souvislé sekvence DNA (33). Při této metodě se vložená DNA amplifikuje pomocí PCR s použitím primerů, které vytvářejí produkt PCR obsahující oblasti překrývající se s vektorem. Vektor a insert se poté smíchají, denaturují a annealují, což umožní hybridizaci insertu s vektorem. Druhým kolem PCR se vytvoří rekombinantní molekuly DNA vektoru obsahujícího insert. PCR s překrývajícím se rozšířením umožnila výzkumníkům spojit dohromady velké geny, které nebylo možné snadno amplifikovat tradičními metodami PCR. Overlap extension PCR se také používala k zavádění mutací do genových sekvencí (34).
Obrázek 2. Přehled PCR

Vývoj specializovaných klonovacích technik.
Ve snaze dále zvýšit účinnost molekulárního klonování bylo vyvinuto několik specializovaných nástrojů a technik, které využívaly vlastnosti jedinečných enzymů.
Klonování TA. Jeden z přístupů využíval vlastnosti Taq DNA polymerázy, první tepelně stabilní polymerázy používané pro PCR. Taq během amplifikace přidává na konec každého produktu PCR jeden nukleotid 3´ dA. Produkt PCR lze snadno podvázat do vektoru, který byl nastříhán a upraven tak, aby obsahoval jednotlivé T zbytky na každém vlákně. Několik společností uvedlo tuto techniku na trh a prodává soupravy obsahující klonovací vektory, které jsou již linearizované a „ocasaté“.
LIC. Klonování nezávislé na ligaci (LIC), jak napovídá jeho název, umožňuje spojování molekul DNA bez přítomnosti DNA ligázy. LIC se běžně provádí pomocí T4 DNA polymerázy, která se používá k vytvoření jednořetězcových převisů DNA o délce >12 nukleotidů jak na linearizované DNA vektoru, tak na inzertu, který má být klonován (35). Po smíchání se vektor a insert žíhají přes dlouhý úsek kompatibilních konců. Délka kompatibilních konců je dostatečná, aby udržela molekulu pohromadě i bez přítomnosti ligázy, a to i během transformace. Po transformaci jsou mezery opraveny in vivo. Existuje několik různých komerčně dostupných produktů pro klonování LIC.USER.
Klonování USER bylo poprvé vyvinuto na počátku 90. let 20. století jako metoda klonování nezávislá na restrikčních enzymech a ligáze (36). V době svého vzniku metoda spočívala v použití primerů PCR, které obsahovaly ~12 nukleotidový 5´ chvost, v němž byly nejméně čtyři deoxythymidinové báze nahrazeny deoxyuridiny. Produkt PCR byl ošetřen uracilglykosidázou DNA (UDG) a endonukleázou VIII, která vyřízne uracilové báze a zanechá 3´ přesah, který lze annealovat na podobně ošetřený vektor. Společnost NEB prodává enzym USER pro klonovací reakce nezávislé na ligáze a restrikčních enzymech.
Budoucí trendy
Molekulární klonování pokročilo od klonování jednoho fragmentu DNA k sestavení více složek DNA do jednoho souvislého úseku DNA. Nové a vznikající technologie se snaží přeměnit klonování na proces, který je stejně jednoduchý jako uspořádání „bloků“ DNA vedle sebe.
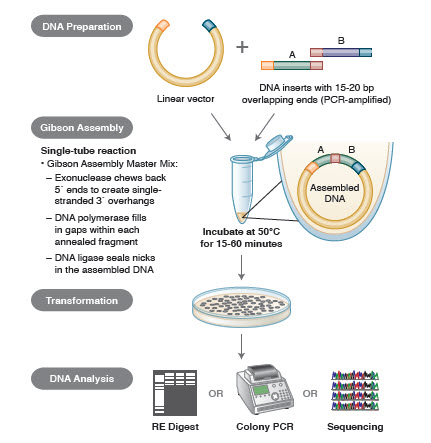
Metody sestavování DNA. Mnoho nových, elegantních technologií umožňuje sestavení více fragmentů DNA v reakci v jedné zkumavce. Výhodou těchto technologií je, že jsou standardizované, bezproblémové a většinou nezávislé na sekvenci. Schopnost sestavit více fragmentů DNA v jedné zkumavce navíc mění sérii dříve nezávislých restrikčních/ligračních reakcí na zjednodušený a efektivní postup.
Různé techniky a produkty pro sestavování genů zahrnují SLIC (Sequence and Ligase Independent Cloning), Gibson Assembly (NEB), GeneArt® Seamless Cloning (Life Technologies) a Gateway® Cloning (Invitrogen) (35,37,38).
Při sestavování DNA jsou bloky DNA, které mají být sestaveny, amplifikovány pomocí PCR. Poté jsou fragmenty DNA, které mají být sestaveny vedle sebe, upraveny tak, aby obsahovaly bloky komplementárních sekvencí, které budou spolu ligovány. Mohou to být kompatibilní kohezivní konce, jako jsou ty, které se používají pro Gibson Assembly, nebo oblasti obsahující rozpoznávací místa pro site-specific rekombinázy (Gateway). Enzym použitý pro ligaci DNA rozpozná a sestaví každou sadu kompatibilních oblastí, čímž se v jedné reakci vytvoří jediná souvislá molekula DNA.
Obrázek 3. Přehled metody klonování Gibson Assembly
Syntetická biologie. Syntéza DNA je oblast syntetické biologie, která v současné době představuje revoluci v technologii rekombinantní DNA. Ačkoli byl poprvé in vitro syntetizován kompletní gen v roce 1972 (40), syntéza velkých molekul DNA se stala realitou až počátkem roku 2000, kdy vědci začali in vitro syntetizovat celé genomy (41,42). Tyto první experimenty trvaly roky, ale technologie urychlují schopnost syntetizovat velké molekuly DNA.
Závěr
Za posledních 40 let se molekulární klonování posunulo od náročné izolace a skládání dvou kousků DNA s následným intenzivním screeningem potenciálních klonů k bezproblémovému sestavení až 10 fragmentů DNA s pozoruhodnou účinností během několika hodin nebo k navrhování molekul DNA in silico a jejich syntéze in vitro. Všechny tyto technologie dohromady poskytují molekulárním biologům úžasně výkonný soubor nástrojů pro zkoumání, manipulaci a využití DNA, který dále rozšíří obzory vědy. Mezi tyto možnosti patří vývoj bezpečnějších rekombinantních proteinů pro léčbu nemocí, zdokonalení genové terapie (43) a rychlejší výroba, ověřování a uvolňování nových vakcín (44). V konečném důsledku je však potenciál omezen pouze naší představivostí.
Rebecca Tirabassi je asistentkou editora na Bitesizebio.com.
Přečtěte si našeho technického průvodce molekulárním klonováním
.