Le clonage moléculaire, un terme qui en est venu à signifier la création de molécules d’ADN recombinant, a stimulé le progrès dans toutes les sciences de la vie. Depuis les années 1970, avec la découverte des endonucléases de restriction – des enzymes qui coupent sélectivement et spécifiquement les molécules d’ADN – la technologie de l’ADN recombinant a connu une croissance exponentielle tant au niveau des applications que de la sophistication, donnant lieu à des outils de plus en plus puissants pour la manipulation de l’ADN. Le clonage des gènes est désormais si simple et si efficace qu’il est devenu une technique de laboratoire standard. Cela a conduit à une explosion de la compréhension de la fonction des gènes au cours des dernières décennies. Les technologies émergentes promettent des possibilités encore plus grandes, comme la possibilité pour les chercheurs d’assembler de manière transparente de multiples fragments d’ADN et de transformer les plasmides résultants en bactéries, en moins de deux heures, ou l’utilisation de cassettes de gènes interchangeables, qui peuvent être facilement déplacées entre différentes constructions, pour maximiser la vitesse et la flexibilité. Dans un avenir proche, le clonage moléculaire verra probablement l’émergence d’un nouveau paradigme, avec les techniques de biologie synthétique qui permettront la synthèse chimique in vitro de toute construction d’ADN spécifiée in silico. Ces avancées devraient permettre une construction et une itération plus rapides des clones d’ADN, accélérant le développement de vecteurs de thérapie génique, de procédés de production de protéines recombinantes et de nouveaux vaccins.
Rebecca Tirabassi, Bitesize Bio.
- Introduction
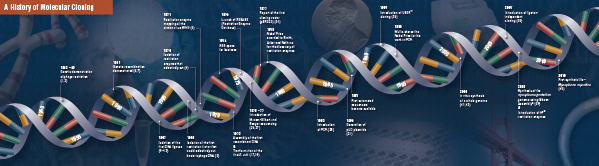
- Histoire du clonage
- Le fondement du clonage moléculaire
- Figure 1. Flux de travail de clonage traditionnel
- Construire sur les bases
- Figure 2. Vue d’ensemble de la PCR
- Tendances futures
- Figure 3. Aperçu de la méthode de clonage par assemblage Gibson
- Conclusion
- Voir notre guide technique du clonage moléculaire
Introduction
Le clonage moléculaire fait référence à l’isolement d’une séquence d’ADN de n’importe quelle espèce (souvent un gène), et à son insertion dans un vecteur pour la propagation, sans altération de la séquence d’ADN originale. Une fois isolés, les clones moléculaires peuvent être utilisés pour générer de nombreuses copies de l’ADN pour l’analyse de la séquence du gène, et/ou pour exprimer la protéine résultante pour l’étude ou l’utilisation de la fonction de la protéine. Les clones peuvent également être manipulés et mutés in vitro pour modifier l’expression et la fonction de la protéine.
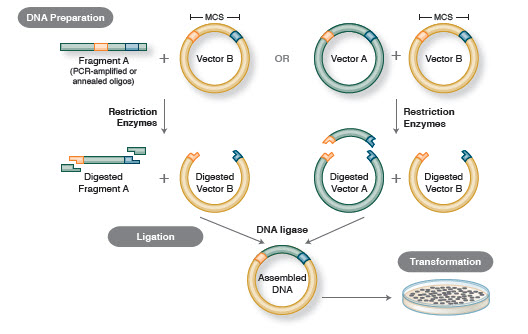
Le flux de travail de base du clonage comprend quatre étapes :
- Isolation de fragments d’ADN cibles (souvent appelés inserts)
- Ligature des inserts dans un vecteur de clonage approprié, créant des molécules recombinantes (par ex, plasmides)
- Transformation des plasmides recombinants dans des bactéries ou d’autres hôtes appropriés pour la propagation
- Criblage/sélection des hôtes contenant le plasmide recombinant prévu
Ces quatre étapes révolutionnaires ont été soigneusement reconstituées et réalisées par de multiples laboratoires, à partir de la fin des années 1960 et du début des années 1970. Un résumé des découvertes qui comprennent le clonage moléculaire traditionnel est décrit dans les pages suivantes.
Histoire du clonage
Le fondement du clonage moléculaire
Découpage (digestion). La technologie de l’ADN recombinant est apparue à la fin des années 1960, avec la découverte d’enzymes capables de couper et de joindre spécifiquement les molécules d’ADN double brin. En fait, dès 1952, deux groupes ont observé indépendamment que les bactéries codaient un « facteur de restriction » qui empêchait les bactériophages de se développer dans certains hôtes (1,2). Mais la nature de ce facteur n’a été découverte qu’en 1968, lorsque Arber et Linn ont réussi à isoler une enzyme, appelée facteur de restriction, qui coupait sélectivement l’ADN exogène, mais pas l’ADN bactérien (3). Ces études ont également identifié une enzyme méthylase qui protégeait l’ADN bactérien des enzymes de restriction.
Peu de temps après la découverte d’Arber et Linn, Smith a étendu et confirmé ces études en isolant une enzyme de restriction de Haemophilus influenza. Il a démontré que l’enzyme coupait sélectivement l’ADN au milieu d’une portion spécifique de 6 paires de bases d’ADN ; une caractéristique de certaines enzymes de restriction est leur propension à couper le substrat d’ADN dans ou près de séquences de « reconnaissance » spécifiques, souvent palindromiques (4).
La pleine puissance des enzymes de restriction n’a été réalisée que lorsque les enzymes de restriction et l’électrophorèse sur gel ont été utilisées pour cartographier le génome du virus simien 40 (SV40) (5). Pour ces découvertes fondamentales, Werner Arber, Hamilton Smith et Daniel Nathans ont partagé le prix Nobel de médecine en 1978.
Figure 1. Flux de travail de clonage traditionnel
Assembling (Ligation). Tout comme la découverte des enzymes qui coupent l’ADN, la découverte d’une enzyme capable d’assembler l’ADN a été précédée d’observations antérieures saillantes. Au début des années 1960, deux groupes ont découvert que la recombinaison génétique pouvait se produire par la rupture et la ligature des molécules d’ADN (6,7), suivie de près par l’observation que l’ADN linéaire du bactériophage est rapidement converti en cercles fermés de manière covalente après l’infection de l’hôte (8). À peine deux ans plus tard, cinq groupes isolent indépendamment des ADN ligases et démontrent leur capacité à assembler deux morceaux d’ADN (9-13).
Peu de temps après la découverte des enzymes de restriction et des ADN ligases, la première molécule d’ADN recombinant a été fabriquée. En 1972, Berg a coupé et ligaturé séparément un morceau d’ADN de bactériophage lambda ou l’opéron galactose de E. coli avec de l’ADN SV40 pour créer les premières molécules d’ADN recombinant (14). Ces études ont ouvert la voie au concept selon lequel, en raison de la nature universelle de l’ADN, l’ADN de n’importe quelle espèce pouvait être assemblé. En 1980, Paul Berg a partagé le prix Nobel de chimie avec Walter Gilbert et Frederick Sanger (les développeurs du séquençage de l’ADN), pour « ses études fondamentales sur la biochimie des acides nucléiques, en particulier en ce qui concerne l’ADN recombinant. »
Transformation. La technologie de l’ADN recombinant serait gravement limitée, et le clonage moléculaire impossible, sans les moyens de propager et d’isoler la molécule d’ADN nouvellement construite. La capacité de transformer des bactéries, ou de provoquer l’absorption, l’incorporation et l’expression de matériel génétique étranger, a été démontrée pour la première fois par Griffith lorsqu’il a transformé une souche non létale de bactéries en une souche létale en mélangeant la souche non létale avec des bactéries létales inactivées par la chaleur (15). Cependant, la nature du « principe de transformation » qui confère la létalité n’a pas été comprise avant 1944. La même année, Avery, Macleod et McCarty ont démontré que l’ADN, et non les protéines, était responsable de l’induction du phénotype létal (16).
A l’origine, on croyait que la souche bactérienne de laboratoire commune, E. coli, était réfractaire à la transformation, jusqu’à ce que Mandel et Higa démontrent que le traitement d’E. coli avec du chlorure de calcium induisait la prise d’ADN de bactériophage (17). Cohen a appliqué ce principe, en 1972, lorsqu’il a été le premier à transformer des bactéries avec des plasmides pour leur conférer une résistance aux antibiotiques (18).
L’expérience ultime : digestion, ligature et transformation d’une molécule d’ADN recombinant a été exécutée par Boyer, Cohen et Chang en 1973, lorsqu’ils ont digéré le plasmide pSC101 avec EcoRI, ligaturé le fragment linéarisé à un autre plasmide restreint par une enzyme et transformé la molécule recombinante résultante dans E. coli, conférant à la bactérie une résistance à la tétracycline (19), posant ainsi les bases de la plupart des travaux sur l’ADN recombinant depuis.
Construire sur les bases
Bien que les scientifiques aient découvert et appliqué tous les principes de base pour créer et propager l’ADN recombinant dans les bactéries, le processus était inefficace. Les préparations d’enzymes de restriction n’étaient pas fiables en raison de procédures de purification non standardisées, les plasmides pour le clonage étaient encombrants, difficiles à travailler et limités en nombre, et les expériences étaient limitées par la quantité d’ADN d’insertion qui pouvait être isolée. Les recherches menées au cours des décennies suivantes ont permis d’améliorer les techniques et les outils disponibles pour le clonage moléculaire.
Première conception de vecteurs.
Développement du premier vecteur standardisé. Les scientifiques travaillant dans le laboratoire de Boyer ont reconnu la nécessité d’un plasmide de clonage général, un plasmide compact avec des sites de restriction uniques pour le clonage d’ADN étranger et l’expression de gènes de résistance aux antibiotiques pour la sélection des bactéries transformées. En 1977, ils ont décrit le premier vecteur conçu à des fins de clonage, pBR322 (20). Ce vecteur était petit, d’une taille de ~4 kilobases, et possédait deux gènes de résistance aux antibiotiques pour la sélection.
Vecteurs avec sélection embarquée et rendements plus élevés. Bien que la sélection antibiotique empêchait les bactéries non transformées de se développer, les plasmides qui se re-ligaturaient sans fragments d’ADN d’insertion (auto-ligature) pouvaient encore conférer une résistance aux antibiotiques aux bactéries. Par conséquent, trouver les bons clones bactériens contenant la molécule d’ADN recombinant souhaitée pouvait prendre beaucoup de temps.
Vieira et Messing ont conçu un outil de dépistage pour identifier les colonies bactériennes contenant des plasmides avec des insertions d’ADN. En se basant sur le plasmide pBR322, ils ont créé la série de plasmides pUC, qui contenait un système de « dépistage bleu/blanc » (21). Le placement d’un site de clonage multiple (MCS) contenant plusieurs sites de restriction uniques dans le gène LacZ’ a permis aux chercheurs de cribler les colonies bactériennes contenant des plasmides avec l’insert d’ADN étranger. Lorsque les bactéries étaient placées sur les milieux appropriés, les colonies blanches contenaient des plasmides avec des inserts, tandis que les colonies bleues contenaient des plasmides sans inserts. Les plasmides pUC présentaient un avantage supplémentaire par rapport aux vecteurs existants ; ils contenaient une mutation qui entraînait un nombre de copies plus élevé, augmentant ainsi les rendements plasmidiques.
Amélioration des digestions de restriction. Les premiers travaux avec les enzymes de restriction ont été entravés par la pureté de la préparation enzymatique et un manque de compréhension des exigences de tampon pour chaque enzyme. En 1975, New England Biolabs (NEB) a été la première société à commercialiser des enzymes de restriction produites à partir d’une source recombinante. Cela a permis d’obtenir des rendements plus élevés, une plus grande pureté, une constance de lot à lot et des prix plus bas. Actuellement, plus de 4 000 enzymes de restriction, reconnaissant plus de 300 séquences différentes, ont été découvertes par des scientifiques du monde entier. NEB fournit actuellement plus de 230 de ces spécificités.
NEB a également été l’une des premières entreprises à développer un système standardisé à quatre tampons, et à caractériser toutes ses activités enzymatiques dans ce système tampon. Cela a permis de mieux comprendre comment réaliser une double digestion, ou la digestion de l’ADN avec deux enzymes simultanément. Des recherches ultérieures ont conduit au développement de systèmes à un tampon, compatibles avec les enzymes de restriction les plus courantes (comme le tampon CutSmart™ de NEB).
Avec l’avènement des bibliothèques d’enzymes de restriction disponibles dans le commerce et dont les spécificités de séquence sont connues, les enzymes de restriction sont devenues un outil puissant pour cribler les clones potentiels d’ADN recombinant. La « digestion diagnostique » était, et est toujours, l’une des techniques les plus courantes utilisées dans le clonage moléculaire.
Préparation du vecteur et de l’insert. L’efficacité et la polyvalence du clonage ont également été améliorées par le développement de différentes techniques de préparation des vecteurs avant la ligature. On a isolé des phosphatases alcalines capables d’éliminer les groupes phosphate 3′ et 5′ des extrémités de l’ADN . On a rapidement découvert que le traitement des vecteurs avec la phosphatase intestinale de veau (CIP) déphosphorylait les extrémités de l’ADN et empêchait l’auto-ligature du vecteur, augmentant la récupération des plasmides avec insert (24).
L’enzyme CIP s’est avérée difficile à inactiver, et toute activité résiduelle a conduit à la déphosphorylation de l’ADN d’insert et à l’inhibition de la réaction de ligature. La découverte des phosphatases alcalines thermolabiles, comme la phosphatase alcaline recombinante de la crevette (rSAP) et la phosphatase antarctique (AP) (toutes deux vendues par NEB), a diminué les étapes et le temps nécessaires, car un simple changement de température inactive l’enzyme avant l’étape de ligature (25).
Le séquençage de l’ADN arrive. Le séquençage de l’ADN a été développé à la fin des années 1970, lorsque deux méthodes concurrentes ont été conçues. Maxam et Gilbert ont développé la « méthode de séquençage chimique », qui reposait sur la modification chimique de l’ADN et le clivage ultérieur au niveau de bases spécifiques (26). Au même moment, Sanger et ses collègues ont publié la « méthode de terminaison de chaîne », qui est devenue la méthode utilisée par la plupart des chercheurs (27). La méthode de Sanger s’est rapidement automatisée et les premiers séquenceurs automatiques ont été vendus en 1987.
La possibilité de déterminer la séquence d’un tronçon d’ADN a renforcé la fiabilité et la polyvalence du clonage moléculaire. Une fois clonés, les scientifiques pouvaient séquencer les clones pour identifier définitivement la bonne molécule recombinante, identifier de nouveaux gènes ou des mutations dans les gènes, et concevoir facilement des oligonucléotides basés sur la séquence connue pour des expériences supplémentaires.
L’impact de la réaction en chaîne par polymérase. L’un des problèmes du clonage moléculaire dans les premières années était d’obtenir suffisamment d’ADN d’insertion pour le cloner dans le vecteur. En 1983, Mullis a conçu une technique qui a résolu ce problème et révolutionné le clonage moléculaire (28). Il a amplifié une portion d’ADN cible en utilisant des amorces opposées pour amplifier les deux brins complémentaires d’ADN, simultanément. Grâce à des cycles de dénaturation, de recuit et de polymérisation, il a montré qu’il pouvait amplifier de manière exponentielle une seule copie d’ADN. La réaction en chaîne par polymérase, ou PCR, a permis d’amplifier et de cloner des gènes à partir de quantités d’ADN auparavant insuffisantes. Pour cette découverte, Kary Mullis a partagé le prix Nobel de chimie de 1993 « pour ses contributions au développement de méthodes dans le cadre de la chimie de l’ADN ».
En 1970, Temin et Baltimore ont découvert indépendamment la transcriptase inverse dans les virus, une enzyme qui convertit l’ARN en ADN (29,30). Peu après le développement de la PCR, la transcription inverse a été couplée à la PCR (RT-PCR) pour permettre le clonage de l’ARN messager (ARNm). La transcription inverse a été utilisée pour créer une copie d’ADN (ADNc) de l’ARNm qui a ensuite été amplifié par PCR pour créer un insert pour la ligature. Pour leur découverte de l’enzyme, Howard Temin et David Baltimore ont reçu le prix Nobel de médecine et de physiologie en 1975, qu’ils ont partagé avec Renato Dulbecco.
Clonage de produits PCR. L’avènement de la PCR signifiait que les chercheurs pouvaient désormais cloner des gènes et des segments d’ADN avec une connaissance limitée de la séquence de l’amplicon. Cependant, il y avait peu de consensus quant à la méthode optimale de préparation des produits PCR pour une ligature efficace dans les vecteurs de clonage.
Plusieurs méthodes différentes ont été initialement utilisées pour cloner les produits PCR. La méthode la plus simple, et encore la plus courante, pour cloner des produits PCR consiste à introduire des sites de restriction aux extrémités du produit PCR (31). Cela permet le clonage direct et directionnel de l’insert dans le vecteur après la digestion de restriction. Le clonage à extrémités émoussées a été développé pour ligaturer directement les produits PCR générés par des polymérases qui produisent des extrémités émoussées, ou des inserts conçus pour avoir des sites de restriction qui laissent des extrémités émoussées une fois l’insert digéré. Ceci était utile pour cloner des fragments d’ADN qui ne contenaient pas de sites de restriction compatibles avec le vecteur (32).
Peu de temps après l’introduction de la PCR, la PCR à extension par chevauchement a été introduite comme méthode pour assembler les produits PCR en une séquence d’ADN contiguë (33). Dans cette méthode, l’insert d’ADN est amplifié par PCR en utilisant des amorces qui génèrent un produit de PCR contenant des régions chevauchantes avec le vecteur. Le vecteur et l’insert sont ensuite mélangés, dénaturés et hybridés, ce qui permet l’hybridation de l’insert au vecteur. Un deuxième cycle de PCR génère des molécules d’ADN recombinant du vecteur contenant l’insert. La PCR à extension par chevauchement a permis aux chercheurs de reconstituer de grands gènes qui ne pouvaient pas être facilement amplifiés par les méthodes de PCR traditionnelles. La PCR à extension par chevauchement a également été utilisée pour introduire des mutations dans des séquences de gènes (34).
Figure 2. Vue d’ensemble de la PCR

Développement de techniques de clonage spécialisées.
Dans un effort pour améliorer encore l’efficacité du clonage moléculaire, plusieurs outils et techniques spécialisés ont été développés qui exploitaient les propriétés d’enzymes uniques.
Clonage TA. Une approche a tiré parti d’une propriété de l’ADN polymérase Taq, la première polymérase thermostable utilisée pour la PCR. Pendant l’amplification, Taq ajoute un seul nucléotide 3′ dA à l’extrémité de chaque produit PCR. Le produit PCR peut être facilement ligaturé dans un vecteur qui a été coupé et modifié pour contenir des résidus T uniques sur chaque brin. Plusieurs sociétés ont commercialisé cette technique et vendent des kits contenant des vecteurs de clonage qui sont déjà linéarisés et « tailed ».
LIC. Le clonage indépendant de la ligature (LIC), comme son nom l’indique, permet de joindre des molécules d’ADN en l’absence d’ADN ligase. Le LIC est généralement réalisé avec l’ADN polymérase T4, qui est utilisée pour générer des surplombs d’ADN simple brin, d’une longueur de >12 nucléotides, à la fois sur l’ADN vecteur linéarisé et sur l’insert à cloner (35). Lorsqu’ils sont mélangés, le vecteur et l’insert s’annèlent par le long tronçon d’extrémités compatibles. La longueur des extrémités compatibles est suffisante pour maintenir la molécule ensemble en l’absence de ligase, même pendant la transformation. Une fois transformées, les lacunes sont réparées in vivo. Il existe plusieurs produits différents disponibles dans le commerce pour le clonage LIC.USER.
Le clonage USER a été développé pour la première fois au début des années 1990 comme une méthode de clonage indépendante des enzymes de restriction et des ligases (36). Lorsqu’elle a été conçue, la méthode reposait sur l’utilisation d’amorces PCR qui contenaient une queue 5′ de ~12 nucléotides, dans laquelle au moins quatre bases de désoxythymidine avaient été substituées par des désoxyuridines. Le produit de la PCR a été traité avec de l’uracile ADN glycosidase (UDG) et de l’endonucléase VIII, ce qui excise les bases uracile et laisse un chevauchement en 3′ qui peut être annelé à un vecteur traité de façon similaire. NEB vend l’enzyme USER pour les réactions de clonage indépendantes des ligases et des enzymes de restriction.
Tendances futures
Le clonage moléculaire a progressé du clonage d’un seul fragment d’ADN à l’assemblage de plusieurs composants d’ADN dans un seul tronçon contigu d’ADN. Les technologies nouvelles et émergentes cherchent à transformer le clonage en un processus aussi simple que la disposition de « blocs » d’ADN les uns à côté des autres.
Méthodes d’assemblage d’ADN. De nombreuses technologies nouvelles et élégantes permettent l’assemblage de multiples fragments d’ADN dans une réaction à un seul tube. Les avantages de ces technologies sont qu’elles sont standardisées, sans rupture et majoritairement indépendantes de la séquence. En outre, la possibilité d’assembler plusieurs fragments d’ADN dans un seul tube transforme une série de réactions de restriction/ligature auparavant indépendantes en une procédure rationalisée et efficace.
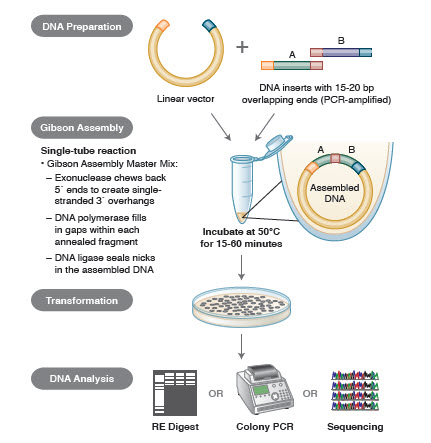
Les différentes techniques et produits pour l’assemblage de gènes incluent SLIC (Sequence and Ligase Independent Cloning), Gibson Assembly (NEB), GeneArt® Seamless Cloning (Life Technologies) et Gateway® Cloning (Invitrogen) (35,37,38).
Dans l’assemblage d’ADN, les blocs d’ADN à assembler sont amplifiés par PCR. Ensuite, les fragments d’ADN à assembler adjacents les uns aux autres sont conçus pour contenir des blocs de séquences complémentaires qui seront ligaturés ensemble. Il peut s’agir d’extrémités cohésives compatibles, comme celles utilisées pour l’assemblage de Gibson, ou de régions contenant des sites de reconnaissance pour les recombinases spécifiques de site (Gateway). L’enzyme utilisée pour la ligature de l’ADN reconnaîtra et assemblera chaque ensemble de régions compatibles, créant ainsi une molécule d’ADN unique et contiguë en une seule réaction.
Figure 3. Aperçu de la méthode de clonage par assemblage Gibson
Biologie synthétique. La synthèse d’ADN est un domaine de la biologie synthétique qui révolutionne actuellement la technologie de l’ADN recombinant. Bien qu’un gène complet ait été synthétisé pour la première fois in vitro en 1972 (40), la synthèse de grandes molécules d’ADN n’est devenue une réalité qu’au début des années 2000, lorsque les chercheurs ont commencé à synthétiser des génomes entiers in vitro (41,42). Ces premières expériences ont nécessité des années de travail, mais la technologie accélère la capacité à synthétiser de grandes molécules d’ADN.
Conclusion
Au cours des 40 dernières années, le clonage moléculaire a progressé, passant de l’isolement et de l’assemblage ardus de deux morceaux d’ADN, suivis d’un criblage intensif des clones potentiels, à l’assemblage transparent de jusqu’à 10 fragments d’ADN avec une efficacité remarquable en quelques heures seulement, ou à la conception de molécules d’ADN in silico et à leur synthèse in vitro. Ensemble, toutes ces technologies donnent aux biologistes moléculaires une boîte à outils étonnamment puissante pour explorer, manipuler et exploiter l’ADN, qui élargira encore les horizons de la science. Parmi les possibilités, citons la mise au point de protéines recombinantes plus sûres pour le traitement des maladies, l’amélioration de la thérapie génique (43) et l’accélération de la production, de la validation et de la diffusion de nouveaux vaccins (44). Mais en fin de compte, le potentiel n’est limité que par notre imagination.
Rebecca Tirabassi est rédactrice adjointe à Bitesizebio.com.
Voir notre guide technique du clonage moléculaire
.