Klonowanie molekularne, termin oznaczający tworzenie rekombinowanych cząsteczek DNA, pobudziło postęp w naukach przyrodniczych. Począwszy od lat 70-tych, wraz z odkryciem endonukleaz restrykcyjnych – enzymów, które selektywnie i specyficznie przecinają cząsteczki DNA – technologia rekombinowanego DNA odnotowała wykładniczy wzrost zarówno w zakresie zastosowań, jak i zaawansowania, dostarczając coraz potężniejszych narzędzi do manipulacji DNA. Klonowanie genów jest obecnie tak proste i wydajne, że stało się standardową techniką laboratoryjną. Doprowadziło to do eksplozji w zrozumieniu funkcji genów w ostatnich dziesięcioleciach. Pojawiające się technologie obiecują jeszcze większe możliwości, takie jak umożliwienie badaczom bezproblemowego łączenia wielu fragmentów DNA i przekształcania powstałych w ten sposób plazmidów w bakterie w czasie krótszym niż dwie godziny, lub stosowanie wymiennych kaset genowych, które można łatwo przenosić między różnymi konstruktami, aby zmaksymalizować szybkość i elastyczność. W niedalekiej przyszłości klonowanie molekularne będzie prawdopodobnie świadkiem pojawienia się nowego paradygmatu, z technikami biologii syntetycznej, które umożliwią chemiczną syntezę in vitro dowolnego konstruktu DNA określonego w krzemie. Postępy te powinny umożliwić szybszą konstrukcję i iterację klonów DNA, przyspieszając rozwój wektorów terapii genowej, procesów produkcji białek rekombinowanych i nowych szczepionek.
Rebecca Tirabassi, Bitesize Bio.
Wprowadzenie
Klonowanie molekularne odnosi się do izolacji sekwencji DNA z dowolnego gatunku (często jest to gen) i jej wstawienia do wektora w celu rozmnażania, bez zmiany oryginalnej sekwencji DNA. Po wyizolowaniu, klony molekularne mogą być wykorzystywane do generowania wielu kopii DNA do analizy sekwencji genu, i/lub do ekspresji powstałego białka do badania lub wykorzystania funkcji białka. Klony mogą być również manipulowane i mutowane in vitro w celu zmiany ekspresji i funkcji białka.
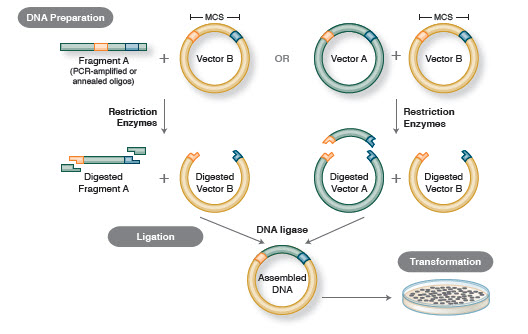
Podstawowy przebieg klonowania obejmuje cztery etapy:
- Isolacja docelowych fragmentów DNA (często określanych jako inserty)
- Ligacja insertów do odpowiedniego wektora klonowania, tworzenie rekombinowanych cząsteczek (np, plazmidy)
- Transformacja rekombinowanych plazmidów do bakterii lub innego odpowiedniego gospodarza w celu rozmnażania
- Badanie przesiewowe/wybór gospodarzy zawierających zamierzony rekombinowany plazmid
Te cztery przełomowe etapy zostały starannie połączone i wykonane przez wiele laboratoriów, począwszy od późnych lat 60. i wczesnych 70. Podsumowanie odkryć, które składają się na tradycyjne klonowanie molekularne, opisano na kolejnych stronach.
Historia klonowania
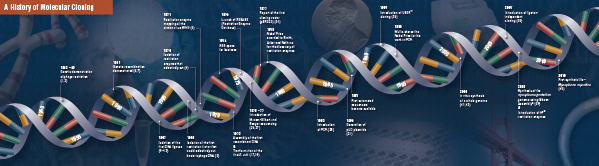
Fundacja klonowania molekularnego
Cięcie (trawienie). Technologia rekombinowanego DNA pojawiła się po raz pierwszy w późnych latach 60-tych, wraz z odkryciem enzymów, które mogły specyficznie ciąć i łączyć dwuniciowe cząsteczki DNA. W rzeczywistości, już w 1952 r. dwie grupy niezależnie zaobserwowały, że bakterie kodują „czynnik restrykcyjny”, który zapobiega rozwojowi bakteriofagów w obrębie niektórych gospodarzy (1,2). Jednak natura tego czynnika nie została odkryta aż do 1968 roku, kiedy to Arberowi i Linnowi udało się wyizolować enzym, nazwany czynnikiem restrykcyjnym, który selektywnie ciął egzogenne DNA, ale nie DNA bakteryjne (3). Badania te zidentyfikowały również enzym metylazę, który chronił bakteryjne DNA przed enzymami restrykcyjnymi.
Wkrótce po odkryciu Arbera i Linna, Smith rozszerzył i potwierdził te badania poprzez wyizolowanie enzymu restrykcyjnego z Haemophilus influenza. Wykazał on, że enzym ten selektywnie przecinał DNA w środku specyficznego odcinka DNA o długości 6 par zasad; jedną z cech charakterystycznych niektórych enzymów restrykcyjnych jest ich skłonność do przecinania substratu DNA w lub w pobliżu specyficznych, często palindromicznych, sekwencji „rozpoznawania” (4).
Pełna moc enzymów restrykcyjnych nie została zrealizowana, dopóki enzymy restrykcyjne i elektroforeza żelowa nie zostały wykorzystane do mapowania genomu wirusa Simian Virus 40 (SV40) (5). Za te przełomowe odkrycia Werner Arber, Hamilton Smith i Daniel Nathans otrzymali w 1978 r. Nagrodę Nobla w dziedzinie medycyny.
Rysunek 1. Tradycyjny proces klonowania
Assembling (Ligation). Podobnie jak odkrycie enzymów, które przecinają DNA, odkrycie enzymu, który może łączyć DNA, było poprzedzone wcześniejszymi, ważnymi obserwacjami. Na początku lat 60-tych dwie grupy odkryły, że rekombinacja genetyczna może zachodzić poprzez łamanie i łączenie cząsteczek DNA (6,7), po czym nastąpiła obserwacja, że liniowe DNA bakteriofagów jest szybko przekształcane w kowalencyjnie zamknięte kręgi po zakażeniu gospodarza (8). Zaledwie dwa lata później pięć grup niezależnie wyizolowało ligazy DNA i zademonstrowało ich zdolność do składania dwóch fragmentów DNA (9-13).
Niedługo po odkryciu enzymów restrykcyjnych i ligaz DNA, pierwsza rekombinowana cząsteczka DNA została wykonana. W 1972 roku Berg osobno wyciął i podwiązał kawałek DNA bakteriofaga lambda lub operonu galaktozy E. coli z DNA SV40, aby stworzyć pierwsze rekombinowane cząsteczki DNA (14). Badania te zapoczątkowały koncepcję, że ze względu na uniwersalną naturę DNA, DNA z dowolnego gatunku może być łączone ze sobą. W 1980 roku Paul Berg otrzymał Nagrodę Nobla w dziedzinie chemii wraz z Walterem Gilbertem i Frederickiem Sangerem (twórcami sekwencjonowania DNA), za „fundamentalne badania biochemii kwasów nukleinowych, ze szczególnym uwzględnieniem rekombinowanego DNA”.
Transformacja. Technologia rekombinacji DNA byłaby poważnie ograniczona, a klonowanie molekularne niemożliwe, bez środków do propagacji i izolacji nowo skonstruowanej cząsteczki DNA. Zdolność do przekształcania bakterii, lub indukowania wchłaniania, włączania i wyrażania obcego materiału genetycznego, została po raz pierwszy zademonstrowana przez Griffitha, kiedy przekształcił on nieśmiertelny szczep bakterii w szczep śmiertelny poprzez zmieszanie szczepu nieśmiertelnego z inaktywowanymi cieplnie bakteriami śmiertelnymi (15). Jednakże natura „zasady transformacji”, która przenosiła śmiertelność, nie została zrozumiana aż do 1944 roku. W tym samym roku Avery, Macleod i McCarty wykazali, że DNA, a nie białko, było odpowiedzialne za wywoływanie śmiertelnego fenotypu (16).
Początkowo sądzono, że wspólny bakteryjny szczep laboratoryjny, E. coli, jest oporny na transformację, dopóki Mandel i Higa nie wykazali, że traktowanie E. coli chlorkiem wapnia indukowało wychwyt bakteriofagowego DNA (17). Cohen zastosował tę zasadę w 1972 r., kiedy był pionierem w transformacji bakterii plazmidami w celu nadania im odporności na antybiotyki (18).
Ostateczny eksperyment: trawienie, ligacja i transformacja rekombinowanej cząsteczki DNA został przeprowadzony przez Boyera, Cohena i Changa w 1973 roku, kiedy to strawili plazmid pSC101 za pomocą EcoRI, podwiązali zlinearyzowany fragment do innego plazmidu z ograniczeniem enzymatycznym i transformowali powstałą rekombinowaną cząsteczkę do E. coli, nadając bakteriom oporność na tetracyklinę (19), kładąc w ten sposób podwaliny pod większość prac nad rekombinowanym DNA od tamtego czasu.
Building on the Groundwork
While naukowcy odkryli i zastosowali wszystkie podstawowe zasady tworzenia i propagacji rekombinowanego DNA w bakteriach, proces ten był nieefektywny. Preparaty enzymów restrykcyjnych były zawodne z powodu niestandaryzowanych procedur oczyszczania, plazmidy do klonowania były kłopotliwe, trudne do pracy i ograniczone w liczbie, a eksperymenty były ograniczone przez ilość insertu DNA, który można było wyizolować. Badania prowadzone w ciągu następnych kilku dekad doprowadziły do udoskonalenia technik i narzędzi dostępnych do klonowania molekularnego.
Wczesne projektowanie wektorów.
Opracowanie pierwszego standaryzowanego wektora. Naukowcy pracujący w laboratorium Boyera dostrzegli potrzebę stworzenia ogólnego plazmidu klonującego, kompaktowego plazmidu z unikalnymi miejscami restrykcyjnymi do klonowania obcego DNA i ekspresji genów oporności na antybiotyki w celu selekcji transformowanych bakterii. W 1977 roku opisali oni pierwszy wektor przeznaczony do klonowania, pBR322 (20). Wektor ten był mały, o rozmiarze ~4 kilobaz, i posiadał dwa geny oporności na antybiotyki do selekcji.
Wektory z przesiewaniem na pokładzie i wyższą wydajnością. Chociaż selekcja antybiotykowa uniemożliwiała wzrost nietransformowanych bakterii, plazmidy, które uległy religacji bez wstawionych fragmentów DNA (samoligacja), nadal mogły nadawać bakteriom oporność na antybiotyki. Dlatego znalezienie właściwych klonów bakteryjnych zawierających pożądaną rekombinowaną cząsteczkę DNA może być czasochłonne.
Vieira i Messing opracowali narzędzie przesiewowe do identyfikacji kolonii bakteryjnych zawierających plazmidy z insercjami DNA. Na podstawie plazmidu pBR322 stworzyli serię plazmidów pUC, które zawierały system „niebieskiego/białego przesiewania” (21). Umieszczenie wielokrotnego miejsca klonowania (MCS) zawierającego kilka unikalnych miejsc restrykcyjnych w obrębie genu LacZ´ umożliwiło badaczom przesiewanie kolonii bakteryjnych zawierających plazmidy z obcą wstawką DNA. Kiedy bakterie były posiewane na odpowiednie podłoża, białe kolonie zawierały plazmidy z wstawkami, podczas gdy niebieskie kolonie zawierały plazmidy bez wstawek. Plazmidy pUC miały dodatkową przewagę nad istniejącymi wektorami; zawierały mutację, która powodowała wyższą liczbę kopii, co zwiększało wydajność plazmidów.
Ulepszanie testów restrykcyjnych. Wczesna praca z enzymami restrykcyjnymi była utrudniona przez czystość preparatu enzymatycznego i brak zrozumienia wymagań buforowych dla każdego enzymu. W 1975 roku New England Biolabs (NEB) stało się pierwszą firmą, która skomercjalizowała enzymy restrykcyjne produkowane z rekombinowanego źródła. Umożliwiło to uzyskanie wyższych wydajności, lepszej czystości, powtarzalności partii i niższych cen. Obecnie ponad 4000 enzymów restrykcyjnych, rozpoznających ponad 300 różnych sekwencji, zostało odkrytych przez naukowców na całym świecie. NEB obecnie dostarcza ponad 230 z tych specyficzności.
NEB była również jedną z pierwszych firm, która opracowała standardowy system czterech buforów i scharakteryzowała wszystkie aktywności swoich enzymów w tym systemie buforowym. Doprowadziło to do lepszego zrozumienia, jak przeprowadzić podwójne trawienie, czyli trawienie DNA dwoma enzymami jednocześnie. Późniejsze badania doprowadziły do opracowania systemów jednobuforowych, które są kompatybilne z najbardziej powszechnymi enzymami restrykcyjnymi (takimi jak CutSmart™ Buffer firmy NEB).
Z chwilą pojawienia się komercyjnie dostępnych bibliotek enzymów restrykcyjnych o znanej specyficzności sekwencji, enzymy restrykcyjne stały się potężnym narzędziem do przesiewania potencjalnych klonów rekombinowanego DNA. Trawienie diagnostyczne” było i nadal jest jedną z najczęstszych technik stosowanych w klonowaniu molekularnym.
Przygotowanie wektorów i insertów. Wydajność i wszechstronność klonowania została również zwiększona poprzez rozwój różnych technik przygotowywania wektorów przed ligacją. Wyizolowano fosfatazy alkaliczne, które mogły usuwać grupy fosforanowe 3´ i 5´ z końców DNA. Wkrótce odkryto, że traktowanie wektorów Calf-Intestinal Phosphatase (CIP) dephosphorylated DNA ends and prevented self-ligation of the vector, increasing recovery of plasmids with insert (24).
The CIP enzyme proved difficult to inactivate, and any residual activity led to dephosphorylation of insert DNA and inhibition of the ligation reaction. Odkrycie termolabilnych fosfataz alkalicznych, takich jak rekombinowana fosfataza alkaliczna z krewetek (rSAP) i fosfataza antarktyczna (AP) (obie sprzedawane przez NEB), zmniejszyło liczbę etapów i skróciło czas reakcji, ponieważ prosta zmiana temperatury unieczynnia enzym przed etapem ligacji (25).
Nadchodzi sekwencjonowanie DNA. Sekwencjonowanie DNA zostało opracowane w późnych latach 70-tych, kiedy to powstały dwie konkurencyjne metody. Maxam i Gilbert opracowali „metodę sekwencjonowania chemicznego”, która opierała się na chemicznej modyfikacji DNA i późniejszym rozszczepianiu przy określonych zasadach (26). W tym samym czasie Sanger i współpracownicy opublikowali „metodę terminacji łańcucha”, która stała się metodą stosowaną przez większość badaczy (27). Metoda Sangera szybko została zautomatyzowana, a pierwsze automatyczne sekwenatory zostały sprzedane w 1987 roku.
Możliwość określenia sekwencji odcinka DNA zwiększyła niezawodność i wszechstronność klonowania molekularnego. Po sklonowaniu, naukowcy mogli sekwencjonować klony, aby ostatecznie zidentyfikować prawidłową cząsteczkę rekombinantu, zidentyfikować nowe geny lub mutacje w genach i łatwo zaprojektować oligonukleotydy na podstawie znanej sekwencji do dodatkowych eksperymentów.
Wpływ łańcuchowej reakcji polimerazy. Jednym z problemów w klonowaniu molekularnym we wczesnych latach było uzyskanie wystarczającej ilości insertowego DNA do sklonowania do wektora. W 1983 r. Mullis opracował technikę, która rozwiązała ten problem i zrewolucjonizowała klonowanie molekularne (28). Amplifikował on odcinek docelowego DNA, używając przeciwstawnych starterów do amplifikacji obu komplementarnych nici DNA, jednocześnie. Poprzez cykle denaturacji, annealingu i polimeryzacji wykazał, że może w sposób wykładniczy amplifikować pojedynczą kopię DNA. Reakcja łańcuchowa polimerazy, czyli PCR, umożliwiła amplifikację i klonowanie genów z wcześniej niewystarczających ilości DNA. Za to odkrycie Kary Mullis otrzymał w 1993 roku Nagrodę Nobla w dziedzinie chemii „za wkład w rozwój metod w chemii opartej na DNA”.
W 1970 roku Temin i Baltimore niezależnie odkryli odwrotną transkryptazę w wirusach, enzym, który przekształca RNA w DNA (29,30). Wkrótce po opracowaniu PCR, odwrotna transkrypcja została połączona z PCR (RT-PCR), aby umożliwić klonowanie RNA posłańca (mRNA). Odwrotna transkrypcja była wykorzystywana do tworzenia kopii DNA (cDNA) mRNA, które było następnie amplifikowane przez PCR w celu utworzenia wstawki do ligacji. Za odkrycie tego enzymu Howard Temin i David Baltimore otrzymali w 1975 roku Nagrodę Nobla w dziedzinie medycyny i fizjologii, którą podzielili z Renato Dulbecco.
Klonowanie produktów PCR. Pojawienie się PCR oznaczało, że badacze mogli teraz klonować geny i segmenty DNA z ograniczoną wiedzą na temat sekwencji amplikonu. Jednakże, nie było zgody co do optymalnej metody przygotowania produktów PCR w celu wydajnej ligacji do wektorów klonujących.
Początkowo do klonowania produktów PCR stosowano kilka różnych metod. Najprostszą i wciąż najbardziej powszechną metodą klonowania produktów PCR jest wprowadzenie miejsc restrykcyjnych na końce produktu PCR (31). Pozwala to na bezpośrednie, kierunkowe klonowanie insertu do wektora po uprzednim trawieniu restrykcyjnym. Klonowanie z tępymi końcami zostało opracowane w celu bezpośredniej ligacji produktów PCR generowanych przez polimerazy, które wytwarzały tępe końce, lub insertów zaprojektowanych tak, aby posiadały miejsca restrykcyjne, które pozostawiały tępe końce po strawieniu insertu. Było to przydatne w klonowaniu fragmentów DNA, które nie zawierały miejsc restrykcyjnych zgodnych z wektorem (32).
Krótko po wprowadzeniu PCR, wprowadzono overlap extension PCR jako metodę łączenia produktów PCR w jedną ciągłą sekwencję DNA (33). W tej metodzie, insert DNA jest amplifikowany przez PCR przy użyciu starterów, które generują produkt PCR zawierający regiony nakładające się z wektorem. Wektor i insert są następnie mieszane, denaturowane i annektowane, co pozwala na hybrydyzację insertu do wektora. Druga runda PCR generuje rekombinowane cząsteczki DNA wektora zawierającego insercję. PCR z rozszerzeniem nakładania umożliwił badaczom łączenie dużych genów, które nie mogły być łatwo amplifikowane za pomocą tradycyjnych metod PCR. Overlap extension PCR był również wykorzystywany do wprowadzania mutacji do sekwencji genów (34).
Rysunek 2. Przegląd PCR

Rozwój specjalistycznych technik klonowania.
W dążeniu do dalszej poprawy wydajności klonowania molekularnego opracowano kilka specjalistycznych narzędzi i technik, które wykorzystywały właściwości unikalnych enzymów.
TA Cloning. Jedno z podejść wykorzystało właściwość polimerazy Taq DNA, pierwszej termostabilnej polimerazy używanej do PCR. Podczas amplifikacji, Taq dodaje pojedynczy nukleotyd 3´ dA do końca każdego produktu PCR. Produkt PCR może być łatwo ligowany do wektora, który został pocięty i zmodyfikowany tak, aby zawierał pojedyncze reszty T na każdej nici. Kilka firm wprowadziło tę technikę na rynek i sprzedaje zestawy zawierające wektory klonujące, które są już zlinearyzowane i „ogoniaste”.
LIC. Klonowanie niezależne od ligacji (LIC), jak sama nazwa wskazuje, pozwala na łączenie cząsteczek DNA przy nieobecności ligazy DNA. LIC jest powszechnie wykonywane przy użyciu polimerazy T4 DNA, która jest używana do generowania jednoniciowych nawisów DNA, o długości >12 nukleotydów, zarówno na linearyzowanym wektorowym DNA, jak i na wstawce, która ma być sklonowana (35). Po zmieszaniu razem, wektor i insercja annealują poprzez długi odcinek zgodnych końców. Długość zgodnych końców jest wystarczająca, aby utrzymać cząsteczkę razem przy braku ligazy, nawet podczas transformacji. Po transformacji, luki są naprawiane in vivo. Istnieje kilka różnych dostępnych w handlu produktów do klonowania LIC.USER.
Klonowanie USER zostało po raz pierwszy opracowane na początku lat 90-tych jako metoda klonowania niezależna od enzymów restrykcyjnych i ligaz (36). W momencie powstania, metoda ta polegała na użyciu primerów PCR zawierających ~12 nukleotydowy 5´ ogon, w którym co najmniej cztery zasady deoksytymidyny zostały zastąpione deoksyurydynami. Produkt PCR był poddawany działaniu glikozydazy uracylowej DNA (UDG) i endonukleazy VIII, która usuwa zasady uracylowe i pozostawia 3´ nakładkę, która może być annealowana do podobnie traktowanego wektora. NEB sprzedaje enzym USER do reakcji klonowania niezależnych od ligaz i enzymów restrykcyjnych.
Przyszłe trendy
Klonowanie molekularne przeszło od klonowania pojedynczego fragmentu DNA do montażu wielu składników DNA w pojedynczym, ciągłym odcinku DNA. Nowe i powstające technologie dążą do przekształcenia klonowania w proces, który jest tak prosty, jak układanie „bloków” DNA obok siebie.
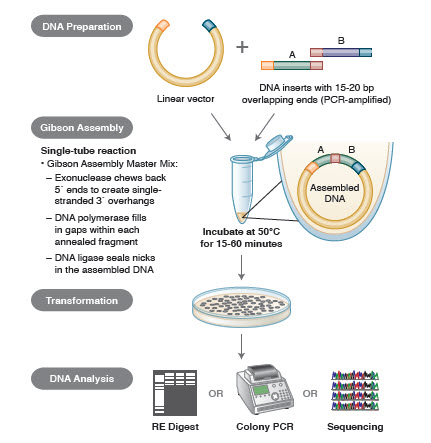
Metody składania DNA. Wiele nowych, eleganckich technologii pozwala na składanie wielu fragmentów DNA w reakcji w jednej probówce. Zaletą tych technologii jest to, że są one standaryzowane, bezproblemowe i w większości przypadków niezależne od sekwencji. Ponadto, zdolność do łączenia wielu fragmentów DNA w jednej probówce zmienia serię wcześniej niezależnych reakcji restrykcji/ligacji w usprawnioną, wydajną procedurę.
Różne techniki i produkty do składania genów obejmują SLIC (Sequence and Ligase Independent Cloning), Gibson Assembly (NEB), GeneArt® Seamless Cloning (Life Technologies) i Gateway® Cloning (Invitrogen) (35,37,38).
W montażu DNA bloki DNA, które mają być złożone, są amplifikowane metodą PCR. Następnie, fragmenty DNA, które mają być złożone obok siebie są zaprojektowane tak, aby zawierały bloki komplementarnych sekwencji, które będą ligowane razem. Mogą to być zgodne końce kohezyjne, takie jak te używane w Gibson Assembly, lub regiony zawierające miejsca rozpoznawania dla rekombinaz specyficznych dla danego miejsca (Gateway). Enzym używany do ligacji DNA rozpozna i złoży każdy zestaw zgodnych regionów, tworząc pojedynczą, przylegającą cząsteczkę DNA w jednej reakcji.
Rysunek 3. Przegląd metody klonowania Gibson Assembly
Biologia syntetyczna. Synteza DNA jest dziedziną biologii syntetycznej, która obecnie rewolucjonizuje technologię rekombinacji DNA. Chociaż kompletny gen został po raz pierwszy zsyntetyzowany in vitro w 1972 roku (40), synteza dużych cząsteczek DNA nie stała się rzeczywistością aż do początku XXI wieku, kiedy to badacze zaczęli syntetyzować całe genomy in vitro (41,42). Te wczesne eksperymenty trwały latami, ale technologia przyspiesza zdolność do syntezy dużych cząsteczek DNA.
Wniosek
W ciągu ostatnich 40 lat klonowanie molekularne przeszło od żmudnego izolowania i łączenia dwóch fragmentów DNA, a następnie intensywnego przesiewania potencjalnych klonów, do bezproblemowego łączenia do 10 fragmentów DNA z niezwykłą wydajnością w ciągu zaledwie kilku godzin, lub projektowania cząsteczek DNA in silico i syntetyzowania ich in vitro. Wszystkie te technologie razem wzięte dają biologom molekularnym zdumiewająco potężny zestaw narzędzi do badania, manipulacji i wykorzystania DNA, który jeszcze bardziej poszerzy horyzonty nauki. Wśród możliwości można wymienić rozwój bezpieczniejszych białek rekombinowanych do leczenia chorób, usprawnienie terapii genowej (43) oraz szybszą produkcję, walidację i wypuszczanie na rynek nowych szczepionek (44). Ale ostatecznie, potencjał jest ograniczony tylko naszą wyobraźnią.
Rebecca Tirabassi jest asystentem redaktora w Bitesizebio.com.