Molekyylikloonaus, jolla tarkoitetaan rekombinantti-DNA-molekyylien luomista, on vauhdittanut kehitystä koko biotieteiden alalla. Rekombinantti-DNA-teknologia alkoi 1970-luvulla restriktioendonukleaasien – entsyymien, jotka leikkaavat DNA-molekyylejä valikoivasti ja spesifisesti – keksimisestä, ja se on kasvanut räjähdysmäisesti sekä sovelluksissa että kehittyneisyydessä, mikä on tuottanut yhä tehokkaampia työkaluja DNA:n manipulointiin. Geenien kloonaus on nykyään niin yksinkertaista ja tehokasta, että siitä on tullut laboratorion vakiotekniikka. Tämä on johtanut geenien toiminnan ymmärtämisen räjähdysmäiseen lisääntymiseen viime vuosikymmeninä. Kehitteillä olevat tekniikat lupaavat vieläkin suurempia mahdollisuuksia, kuten sen, että tutkijat voivat yhdistää saumattomasti useita DNA-fragmentteja ja muuntaa tuloksena syntyneitä plasmideja bakteereihin alle kahdessa tunnissa, tai sen, että nopeuden ja joustavuuden maksimoimiseksi voidaan käyttää vaihtokelpoisia geenikasetteja, joita voidaan helposti siirtää eri konstruktioiden välillä. Lähitulevaisuudessa molekyylikloonaukseen syntyy todennäköisesti uusi paradigma synteettisen biologian tekniikoilla, jotka mahdollistavat minkä tahansa silikonissa määritellyn DNA-konstruktion kemiallisen synteesin in vitro. Nämä edistysaskeleet mahdollistaisivat DNA-kloonien nopeamman rakentamisen ja iteroinnin, mikä nopeuttaisi geeniterapiavektorien, rekombinanttiproteiinien tuotantoprosessien ja uusien rokotteiden kehittämistä.
Rebecca Tirabassi, Bitesize Bio.
Esittely
Molekulaarisella kloonauksella tarkoitetaan DNA-sekvenssin eristämistä miltä tahansa lajilta (usein geenin) ja sen lisäämistä vektoriin lisääntymistä varten alkuperäistä DNA-jaksoa muuttamatta. Kun molekyylikloonit on eristetty, niitä voidaan käyttää tuottamaan useita kopioita DNA:sta geenisekvenssin analysoimiseksi ja/tai tuloksena syntyvän proteiinin ilmentämiseksi proteiinin toiminnan tutkimiseksi tai hyödyntämiseksi. Klooneja voidaan myös manipuloida ja mutatoida in vitro proteiinin ilmentymisen ja toiminnan muuttamiseksi.
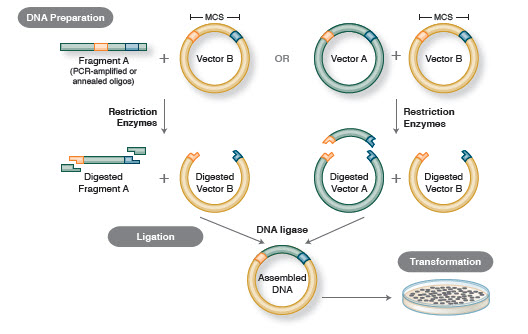
Kloonauksen perustyönkulkuun kuuluu neljä vaihetta:
- Kohteena olevien DNA-fragmenttien (joita usein kutsutaan inserteiksi)
- Inserttien ligaatio sopivaan kloonausvektoriin, jolloin syntyy rekombinanttimolekyylejä (esim, plasmidit)
- Rekombinanttiplasmidien transformaatio bakteereihin tai muuhun sopivaan isäntäkasvattamoon lisääntymistä varten
- Tarkoituksenmukaista rekombinanttiplasmidia sisältävien isäntien seulonta/valinta
Nämä neljä uraauurtavaa vaihetta koottiin huolella yhteen, ja ne suoritettiin useissa eri laboratorioissa 1960-luvun loppupuolelta 1970-luvun alkupuolelta lähtien. Seuraavilla sivuilla kuvataan yhteenveto löydöistä, joista perinteinen molekyylikloonaus koostuu.
Kloonauksen historia
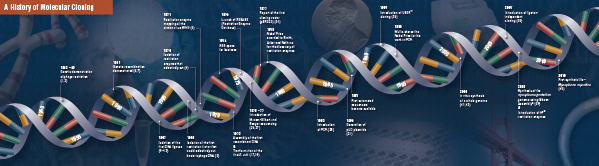
Molekyylikloonauksen perusta
Leikkaus (digestio). Rekombinantti-DNA-teknologia syntyi ensimmäisen kerran 1960-luvun lopulla, kun löydettiin entsyymejä, jotka pystyivät spesifisesti leikkaamaan ja yhdistämään kaksijuosteisia DNA-molekyylejä. Itse asiassa jo vuonna 1952 kaksi ryhmää havaitsi toisistaan riippumatta, että bakteerit koodasivat ”rajoitustekijää”, joka esti bakteriofageja kasvamasta tietyissä isännissä (1,2). Tekijän luonne saatiin kuitenkin selville vasta vuonna 1968, kun Arber ja Linn onnistuivat eristämään restriktiotekijäksi kutsutun entsyymin, joka leikkasi valikoivasti eksogeenista DNA:ta mutta ei bakteerien DNA:ta (3). Näissä tutkimuksissa tunnistettiin myös metylaasientsyymi, joka suojasi bakteeri-DNA:ta restriktioentsyymeiltä.
Lyhyesti Arberin ja Linnin löydön jälkeen Smith laajensi ja vahvisti näitä tutkimuksia eristämällä restriktioentsyymin Haemophilus influenzasta. Hän osoitti, että entsyymi leikkasi DNA:ta valikoivasti tietyn 6 emäsparin pituisen DNA-jakson keskeltä; eräs tiettyjen restriktioentsyymien ominaispiirre on niiden taipumus leikata DNA:n substraattia tietyissä, usein palindromaattisissa ”tunnistussekvensseissä” tai niiden lähellä (4).
Rajoitusentsyymien täysi teho tajuttiin vasta, kun restriktioentsyymejä ja geelielektroforeesia käytettiin Simian Virus 40:n (SV40) genomin kartoittamiseen (5). Näistä uraauurtavista löydöksistä Werner Arber, Hamilton Smith ja Daniel Nathans jakoivat vuoden 1978 lääketieteen Nobel-palkinnon.
Kuva 1. Perinteinen kloonauksen työnkulku
Assembling (Ligation). Aivan kuten DNA:ta leikkaavien entsyymien löytämistä, myös DNA:n yhdistämiseen kykenevän entsyymin löytämistä edelsivät aiemmat, merkittävät havainnot. Kaksi ryhmää havaitsi 1960-luvun alussa, että geneettinen rekombinaatio voi tapahtua DNA-molekyylien katkeamisen ja ligaation kautta (6,7), mitä seurasi läheisesti havainto siitä, että lineaarinen bakteriofagi-DNA muuttuu nopeasti kovalenttisesti suljetuiksi ympyröiksi isännän infektoinnin jälkeen (8). Vain kaksi vuotta myöhemmin viisi ryhmää eristi toisistaan riippumatta DNA-ligaaseja ja osoitti niiden kyvyn yhdistää kaksi DNA-kappaletta (9-13).
Pian restriktioentsyymien ja DNA-ligaasien löytämisen jälkeen valmistettiin ensimmäinen rekombinantti-DNA-molekyyli. Vuonna 1972 Berg leikkasi ja ligoi erikseen palan lambda-bakteriofagin DNA:ta tai E. colin galaktoosio-operonia SV40-DNA:n kanssa luodakseen ensimmäiset rekombinantti-DNA-molekyylit (14). Nämä tutkimukset olivat uraauurtavia siinä, että DNA:n universaalin luonteen vuoksi minkä tahansa lajin DNA:ta voidaan yhdistää toisiinsa. Vuonna 1980 Paul Berg jakoi kemian Nobel-palkinnon Walter Gilbertin ja Frederick Sangerin (DNA:n sekvensoinnin kehittäjät) kanssa ”hänen perustavanlaatuisista tutkimuksistaan nukleiinihappojen biokemiasta ja erityisesti rekombinantti-DNA:sta”.
Transformaatio. Rekombinantti-DNA-teknologia olisi hyvin rajallista ja molekyylikloonaus mahdotonta ilman keinoja, joilla voitaisiin levittää ja eristää vasta rakennettua DNA-molekyyliä. Griffith osoitti ensimmäisen kerran kykynsä transformoida bakteereja eli saada aikaan vieraan geneettisen materiaalin hyväksikäyttöä, sisällyttämistä ja ilmentymistä, kun hän transformoi ei-tappavan bakteerikannan tappavaksi kannaksi sekoittamalla ei-tappavan kannan lämpöinaktivoituihin tappaviin bakteereihin (15). Kuolettavuuden aikaansaavan ”muuntumisperiaatteen” luonne ymmärrettiin kuitenkin vasta vuonna 1944. Samana vuonna Avery, Macleod ja McCarty osoittivat, että DNA, eikä proteiini, oli vastuussa tappavan fenotyypin aikaansaamisesta (16).
Aluksi uskottiin, että yleinen bakteerilaboratoriokanta E. coli oli vastustuskykyinen transformaatiolle, kunnes Mandel ja Higa osoittivat, että kalsiumkloridikäsittely kalsiumkloridilla indusoi bakteriofaagi-DNA:n ottamisen (17). Cohen sovelsi tätä periaatetta vuonna 1972, kun hän teki uraauurtavaa työtä bakteerien muuntamisessa plasmideilla antibioottiresistenssin antamiseksi bakteereille (18).
Viimeisimmän kokeen: rekombinantti-DNA-molekyylin pilkkomisen, ligaation ja transformaation suorittivat Boyer, Cohen ja Chang vuonna 1973, kun he pilkkoivat plasmidin pSC101:n EcoRI:llä, ligaattivat linearisoidun pätkän toiseen entsyymirajoitettuun plasmidiin ja transformoivat tuloksena syntyneen rekombinantti-molekyylin E. coliin ja antoivat bakteerille tetrasykliiniresistenssin (19), mikä loi perustan suurimmalle osalle siitä lähtien tehdyistä rekombinantti-DNA-töistä.
Pohjatyön jatkaminen
Tutkijat olivat keksineet ja soveltaneet kaikkia perusperiaatteita rekombinantti-DNA:n luomiseksi ja levittämiseksi bakteereissa, mutta prosessi ei ollut tehokas. Restriktioentsyymivalmisteet olivat epäluotettavia standardoimattomien puhdistusmenetelmien vuoksi, kloonaukseen käytettävät plasmidit olivat hankalia, vaikeasti työstettäviä ja määrältään rajallisia, ja kokeita rajoitti eristettävän insertti-DNA:n määrä. Seuraavien vuosikymmenten tutkimus johti parannuksiin molekyylikloonaukseen käytettävissä olevissa tekniikoissa ja välineissä.
Varhainen vektorien suunnittelu.
Ensimmäisen standardoidun vektorin kehittäminen. Boyerin laboratoriossa työskentelevät tutkijat havaitsivat, että tarvittiin yleinen kloonausplasmidi, kompakti plasmidi, jossa oli ainutlaatuisia restriktiokohtia vieraan DNA:n kloonausta ja antibioottiresistenssigeenien ilmentämistä varten muunnettujen bakteerien valintaa varten. Vuonna 1977 he kuvasivat ensimmäisen kloonaustarkoituksiin suunnitellun vektorin, pBR322:n (20). Tämä vektori oli pieni, kooltaan ~4 kilobaasia, ja siinä oli kaksi antibioottiresistenssigeeniä valintaa varten.
Vektorit, joissa on mukana seulonta ja suuremmat tuotokset. Vaikka antibioottivalinta esti ei-transformoitujen bakteerien kasvamisen, plasmidit, jotka liukenivat uudelleen ilman insertin DNA-fragmentteja (itseliukioituminen), saattoivat silti antaa bakteereille antibioottiresistenssin. Siksi oikeiden bakteerikloonien löytäminen, jotka sisälsivät halutun rekombinantti-DNA-molekyylin, saattoi olla aikaa vievää.
Vieira ja Messing kehittivät seulontatyökalun, jolla voitiin tunnistaa bakteeripesäkkeet, jotka sisälsivät plasmideja, joissa oli DNA-inserttejä. He loivat pBR322-plasmidin pohjalta sarjan pUC-plasmideja, jotka sisälsivät ”sinivalkoisen seulontajärjestelmän” (21). Moninkertaisen kloonauskohdan (multiple cloning site, MCS), joka sisältää useita ainutlaatuisia restriktiokohtia LacZ´-geenin sisällä, avulla tutkijat pystyivät seulomaan bakteeripesäkkeitä, jotka sisälsivät plasmideja, joissa oli vieras DNA-insertti. Kun bakteerit istutettiin oikeille väliaineille, valkoiset pesäkkeet sisälsivät insertillä varustettuja plasmideja, kun taas siniset pesäkkeet sisälsivät plasmideja, joissa ei ollut inserttiä. pUC-plasmideilla oli lisäetu olemassa oleviin vektoreihin nähden; ne sisälsivät mutaation, joka johti korkeampaan kopiolukuun, mikä lisäsi plasmidien saantoa.
Rajoitussulattujen digestointien parantaminen. Alkuaikojen työtä restriktioentsyymien kanssa haittasivat entsyymivalmisteen puhtaus ja se, ettei kunkin entsyymin puskurivaatimuksia ymmärretty. Vuonna 1975 New England Biolabs (NEB) oli ensimmäinen yritys, joka kaupallisti rekombinanttilähteestä tuotettuja restriktioentsyymejä. Tämä mahdollisti suuremmat saannot, paremman puhtauden, eräkohtaisen yhdenmukaisuuden ja alhaisemman hinnan. Tällä hetkellä tutkijat ympäri maailmaa ovat löytäneet yli 4000 restriktioentsyymiä, jotka tunnistavat yli 300 eri sekvenssiä. NEB toimittaa tällä hetkellä yli 230 näistä spesifikaatioista.
NEB oli myös yksi ensimmäisistä yrityksistä, joka kehitti standardoidun neljän puskurin järjestelmän ja karakterisoi kaikki entsyymiaktiivisuutensa tässä puskurijärjestelmässä. Tämä johti parempaan ymmärrykseen siitä, miten kaksoisdigestiä eli DNA:n pilkkomista kahdella entsyymillä samanaikaisesti voidaan tehdä. Myöhemmin tehdyt tutkimukset johtivat yhden puskurin järjestelmien kehittämiseen, jotka ovat yhteensopivia yleisimpien restriktioentsyymien kanssa (kuten NEB:n CutSmart™-puskuri).
Kun markkinoille tuli kaupallisesti saatavilla olevia restriktioentsyymikirjastoja, joiden sekvenssispesifisyydet ovat tunnettuja, restriktioentsyymeistä tuli tehokas väline potentiaalisten rekombinantti-DNA-kloonien seulontaan. ”Diagnostinen digesti” oli ja on edelleen yksi yleisimmistä molekyylikloonauksessa käytetyistä tekniikoista.
Vektorin ja insertin valmistelu. Kloonauksen tehokkuutta ja monipuolisuutta parannettiin myös kehittämällä erilaisia tekniikoita vektorien valmistamiseksi ennen ligaatiota. Eristettiin emäksisiä fosfataaseja, jotka pystyivät poistamaan 3´ ja 5´ fosfaattiryhmät DNA:n päistä . Pian havaittiin, että vektoreiden käsittely vasikan suolistofosfataasilla (Calf-Intestinal Phosphatase, CIP) defosforyloi DNA:n päät ja esti vektorin itseligoitumisen, mikä lisäsi insertin sisältävien plasmidien talteenottoa (24).
CIP-entsyymiä osoittautui hankalaksi inaktivoida, ja mahdollinen jäljelle jäävä aktiivisuus johti insertti-DNA:n defosforylaatioon ja ligointireaktion estämiseen. Lämpölabiilien emäksisten fosfataasien, kuten rekombinanttisen katkaravun emäksisen fosfataasin (rSAP) ja antarktisen fosfataasin (AP) (molempia myy NEB), keksiminen vähensi työvaiheita ja -aikaa, sillä pelkkä lämpötilan siirto inaktivoi entsyymin ennen ligaatiovaihetta (25).
DNA:n sekvensointi saapuu. DNA:n sekvensointi kehitettiin 1970-luvun lopulla, jolloin kehitettiin kaksi kilpailevaa menetelmää. Maxam ja Gilbert kehittivät ”kemiallisen sekvensointimenetelmän”, joka perustui DNA:n kemialliseen muokkaamiseen ja sitä seuraavaan pilkkomiseen tietyissä emäksissä (26). Samaan aikaan Sanger ja kollegat julkaisivat ”ketjujen lopetusmenetelmän”, josta tuli useimpien tutkijoiden käyttämä menetelmä (27). Sangerin menetelmästä tuli nopeasti automatisoitu, ja ensimmäiset automaattiset sekvensointilaitteet tulivat myyntiin vuonna 1987.
Kyky määrittää DNA-jakson sekvenssi lisäsi molekyylikloonauksen luotettavuutta ja monipuolisuutta. Kun kloonaus oli tehty, tutkijat pystyivät sekvensoimaan kloonit tunnistamaan lopullisesti oikean rekombinanttimolekyylin, tunnistamaan uusia geenejä tai mutaatioita geeneissä ja suunnittelemaan helposti oligonukleotideja tunnetun sekvenssin perusteella lisäkokeita varten.
Polymeraasiketjureaktion vaikutus. Yksi molekyylikloonauksen alkuvuosien ongelmista oli saada riittävästi insertti-DNA:ta kloonattavaksi vektoriin. Vuonna 1983 Mullis kehitti tekniikan, joka ratkaisi tämän ongelman ja mullisti molekyylikloonauksen (28). Hän monisti kohde-DNA:n pätkän käyttämällä vastakkaisia alukkeita, jotka monistivat molemmat komplementaariset DNA-juosteet samanaikaisesti. Denaturointi-, hehkutus- ja polymerointisyklien avulla hän osoitti voivansa monistaa eksponentiaalisesti yhden DNA-kopion. Polymeraasiketjureaktio eli PCR mahdollisti geenien monistamisen ja kloonaamisen aiemmin riittämättömistä DNA-määristä. Tästä keksinnöstä Kary Mullis jakoi vuoden 1993 kemian Nobel-palkinnon ”panoksesta DNA-pohjaisen kemian menetelmien kehittämiseen”.
Vuonna 1970 Temin ja Baltimore löysivät itsenäisesti viruksissa esiintyvän käänteisen transkriptaasin, entsyymin, joka muuntaa RNA:ta DNA:ksi (29,30). Pian PCR:n kehittämisen jälkeen käänteinen transkriptio yhdistettiin PCR:ään (RT-PCR), jotta lähetin-RNA:n (mRNA) kloonaus olisi mahdollista. Käänteistä transkriptiota käytettiin mRNA:n DNA-kopion (cDNA) luomiseen, joka monistettiin myöhemmin PCR:llä insertin luomiseksi ligointia varten. Entsyymin keksimisestä Howard Temin ja David Baltimore saivat vuoden 1975 lääketieteen ja fysiologian Nobel-palkinnon, jonka he jakoivat Renato Dulbeccon kanssa.
PCR-tuotteiden kloonaus. PCR:n tulo merkitsi sitä, että tutkijat pystyivät nyt kloonaamaan geenejä ja DNA-segmenttejä, vaikka amplikonien sekvenssi oli vain vähän tiedossa. PCR-tuotteen valmistuksen optimaalisesta menetelmästä tehokkaan kloonausvektoreihin ligaation mahdollistamiseksi ei kuitenkaan vallinnut yksimielisyyttä.
PCR-tuotteiden kloonaukseen käytettiin aluksi useita erilaisia menetelmiä. Yksinkertaisin ja edelleen yleisin PCR-tuotteiden kloonausmenetelmä on rajoituskohtien lisääminen PCR-tuotteen päihin (31). Tämä mahdollistaa insertin suoran, suunnatun kloonauksen vektoriin restriktiomädätyksen jälkeen. Blunt-ended-kloonaus kehitettiin sellaisten polymeraasien tuottamien PCR-tuotteiden suoraa ligatointia varten, jotka tuottavat tylppiä päitä, tai sellaisten inserttien ligatointia varten, joissa on restriktiokohdat, jotka jättävät tylpät päät, kun insertti on sulatettu. Tästä oli hyötyä kloonattaessa DNA-fragmentteja, jotka eivät sisältäneet vektorin kanssa yhteensopivia restriktiokohtia (32).
Lyhyt aika PCR:n käyttöönoton jälkeen otettiin käyttöön päällekkäispidennyspituus PCR menetelmänä, jolla PCR-tuotteet koottiin yhdeksi yhtenäiseksi DNA-sekvenssiksi (33). Tässä menetelmässä DNA-insertti monistetaan PCR:llä käyttäen alukkeita, jotka tuottavat PCR-tuotteen, joka sisältää vektorin kanssa päällekkäisiä alueita. Tämän jälkeen vektori ja insertti sekoitetaan, denaturoidaan ja yhdistetään, jolloin insertti hybridisoituu vektoriin. Toinen PCR-kierros tuottaa rekombinantti-DNA-molekyylejä insertin sisältävästä vektorista. Overlap extension PCR mahdollisti sen, että tutkijat pystyivät yhdistämään suuria geenejä, joita ei voitu helposti monistaa perinteisillä PCR-menetelmillä. Overlap extension PCR:ää käytettiin myös mutaatioiden tuomiseen geenisekvensseihin (34).
Kuva 2. Yleiskatsaus PCR:ään

Erikoistuneiden kloonaustekniikoiden kehittäminen.
Pyrkimyksenä parantaa edelleen molekyylikloonauksen tehokkuutta kehitettiin useita erikoistuneita työkaluja ja tekniikoita, joissa hyödynnettiin ainutlaatuisten entsyymien ominaisuuksia.
TA-kloonaus. Eräässä lähestymistavassa hyödynnettiin Taq-dna-polymeraasin, ensimmäisen PCR:ssä käytetyn lämpöstabiilin polymeraasin, ominaisuutta. Monistuksen aikana Taq lisää jokaisen PCR-tuotteen loppuun yhden 3´ dA-nukleotidin. PCR-tuote voidaan helposti liittää vektoriin, joka on leikattu ja muokattu siten, että se sisältää yksittäisiä T-jäännöksiä kummassakin säikeessä. Useat yritykset ovat markkinoineet tätä tekniikkaa ja myyvät sarjoja, jotka sisältävät kloonausvektoreita, jotka on jo valmiiksi linearisoitu ja ”pyrstöistetty”.
LIC. Ligation independent cloning (LIC) mahdollistaa nimensä mukaisesti DNA-molekyylien yhdistämisen ilman DNA-ligaasia. LIC suoritetaan yleensä T4-DNA-polymeraasilla, jota käytetään tuottamaan yksisäikeisiä, >12 nukleotidin pituisia DNA-ylijatkoksia sekä linearisoidun vektorin DNA:n että kloonattavan insertin päälle (35). Kun vektori ja insertti sekoitetaan keskenään, vektori ja insertti anneenoituvat pitkien yhteensopivien päiden kautta. Yhteensopivien päiden pituus riittää pitämään molekyylin kasassa ilman ligaasia myös transformaation aikana. Transformoinnin jälkeen aukot korjataan in vivo. LIC.USER-kloonaukseen on useita erilaisia kaupallisesti saatavilla olevia tuotteita.
USER-kloonaus kehitettiin ensimmäisen kerran 1990-luvun alussa restriktioentsyymistä ja ligaasista riippumattomaksi kloonausmenetelmäksi (36). Kun menetelmä alun perin kehitettiin, se perustui sellaisten PCR-alukkeiden käyttöön, jotka sisälsivät ~12 nukleotidin 5´:n hännän, jossa vähintään neljä deoksitymidiiniemästä oli korvattu deoksiuridiinilla. PCR-tuote käsiteltiin urasiili-DNA-glykosidaasilla (UDG) ja endonukleaasi VIII:lla, joka poisti urasiiliemäkset ja jätti jäljelle 3´:n päällekkäisyyden, joka voitiin liittää vastaavalla tavalla käsiteltyyn vektoriin. NEB myy USER-entsyymiä ligaasi- ja restriktioentsyymistä riippumattomia kloonausreaktioita varten.
Tulevaisuuden suuntaukset
Molekulaarinen kloonaus on edennyt yksittäisen DNA-fragmentin kloonauksesta useiden DNA-komponenttien kokoamiseen yhdeksi yhtenäiseksi DNA-jaksoksi. Uusilla ja kehitteillä olevilla tekniikoilla pyritään muuttamaan kloonaus prosessiksi, joka on yhtä yksinkertainen kuin DNA:n ”lohkojen” järjestäminen vierekkäin.
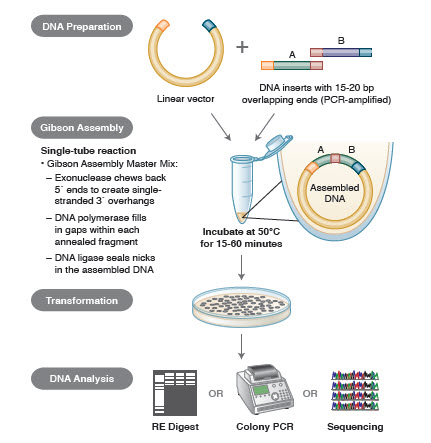
DNA:n kokoamismenetelmät. Monet uudet, tyylikkäät teknologiat mahdollistavat useiden DNA-fragmenttien kokoamisen yhden putken reaktiossa. Näiden tekniikoiden etuna on, että ne ovat standardoituja, saumattomia ja enimmäkseen sekvenssistä riippumattomia. Lisäksi kyky koota useita DNA-fragmentteja yhdessä putkessa muuttaa aiemmin toisistaan riippumattomien restriktio-/ligaatioreaktioiden sarjan virtaviivaiseksi ja tehokkaaksi menettelyksi.
Erilaisia tekniikoita ja tuotteita geenien kokoamiseen ovat SLIC (Sequence and Ligase Independent Cloning), Gibson Assembly (NEB), GeneArt® Seamless Cloning (Life Technologies) ja Gateway® Cloning (Invitrogen) (35,37,38).
DNA-kokoonpanossa koottavat DNA-lohkot monistetaan PCR:llä. Tämän jälkeen vierekkäin koottavat DNA-fragmentit muokataan siten, että ne sisältävät toisiaan täydentäviä sekvenssejä sisältäviä lohkoja, jotka ligmatoidaan yhteen. Nämä voivat olla yhteensopivia koheesiopäätteitä, kuten Gibsonin assemblaatiossa käytettävät, tai alueita, jotka sisältävät tunnistuskohtia paikkaspesifisille rekombinaaseille (Gateway). DNA:n ligaatioon käytettävä entsyymi tunnistaa ja kokoaa kunkin yhteensopivien alueiden joukon, jolloin yhdessä reaktiossa syntyy yksi yhtenäinen DNA-molekyyli.
Kuva 3. Yleiskatsaus Gibsonin assembly-kloonausmenetelmään
Synteettinen biologia. DNA-synteesi on synteettisen biologian osa-alue, joka on parhaillaan mullistamassa rekombinantti-DNA-teknologiaa. Vaikka kokonainen geeni syntetisoitiin ensimmäisen kerran in vitro vuonna 1972 (40), suurten DNA-molekyylien DNA-synteesistä tuli todellisuutta vasta 2000-luvun alussa, jolloin tutkijat alkoivat syntetisoida kokonaisia genomeja in vitro (41,42). Näiden ensimmäisten kokeiden suorittaminen kesti vuosia, mutta tekniikka nopeuttaa kykyä syntetisoida suuria DNA-molekyylejä.
Johtopäätös
Viimeisten 40 vuoden aikana molekyylikloonaus on edennyt kahden DNA-kappaleen vaivalloisesta eristämisestä ja yhdistämisestä, jota seurasi potentiaalisten kloonien intensiivinen seulonta, saumattomaan jopa 10 DNA-fragmentin saumattomaan kokoamiseen huomattavalla tehokkuudella muutamassa tunnissa tai DNA-molekyylien suunnitteluun in silico ja niiden syntetisointiin in vitro. Kaikki nämä tekniikat yhdessä antavat molekyylibiologeille hämmästyttävän tehokkaan työkalupakin DNA:n tutkimiseen, manipulointiin ja hyödyntämiseen, mikä laajentaa tieteen näköaloja entisestään. Mahdollisuuksia ovat muun muassa turvallisempien rekombinanttiproteiinien kehittäminen sairauksien hoitoon, geeniterapian tehostaminen (43) ja uusien rokotteiden nopeampi tuotanto, validointi ja levittäminen (44). Mutta viime kädessä mahdollisuuksia rajoittaa vain mielikuvituksemme.
Rebecca Tirabassi on Bitesizebio.comin apulaistoimittaja.
Katso molekyylikloonauksen tekninen opas
.