Das molekulare Klonen, ein Begriff, der für die Herstellung rekombinanter DNA-Moleküle steht, hat den Fortschritt in den Biowissenschaften vorangetrieben. Seit der Entdeckung der Restriktionsendonukleasen in den 1970er Jahren – Enzyme, die selektiv und spezifisch DNA-Moleküle schneiden – hat die Technologie der rekombinanten DNA ein exponentielles Wachstum sowohl in der Anwendung als auch in der Raffinesse erfahren und immer leistungsfähigere Werkzeuge zur DNA-Manipulation hervorgebracht. Das Klonen von Genen ist heute so einfach und effizient, dass es zu einer Standard-Labortechnik geworden ist. Dies hat in den letzten Jahrzehnten zu einem explosionsartigen Anstieg des Verständnisses der Genfunktionen geführt. Neue Technologien versprechen noch größere Möglichkeiten, wie z. B. die Möglichkeit, mehrere DNA-Fragmente nahtlos zusammenzufügen und die daraus resultierenden Plasmide in weniger als zwei Stunden in Bakterien zu transformieren, oder die Verwendung von austauschbaren Genkassetten, die leicht zwischen verschiedenen Konstrukten ausgetauscht werden können, um die Geschwindigkeit und Flexibilität zu maximieren. In naher Zukunft wird sich beim molekularen Klonen wahrscheinlich ein neues Paradigma herausbilden, mit Techniken der synthetischen Biologie, die eine chemische In-vitro-Synthese jedes beliebigen, in silico spezifizierten DNA-Konstrukts ermöglichen werden. Diese Fortschritte dürften eine schnellere Konstruktion und Iteration von DNA-Klonen ermöglichen und die Entwicklung von Gentherapievektoren, rekombinanten Proteinproduktionsverfahren und neuen Impfstoffen beschleunigen.
Rebecca Tirabassi, Bitesize Bio.
- Einführung
- Geschichte des Klonens
- Die Grundlagen des molekularen Klonens
- Abbildung 1. Traditioneller Klonierungs-Workflow
- Auf den Grundlagen aufbauen
- Abbildung 2. Überblick über die PCR
- Zukunftstrends
- Abbildung 3. Überblick über die Gibson Assembly Klonierungsmethode
- Schlussfolgerung
- Besichtigen Sie unseren technischen Leitfaden zum molekularen Klonen
Einführung
Molekulares Klonen bezieht sich auf die Isolierung einer DNA-Sequenz aus einer beliebigen Spezies (häufig ein Gen) und deren Einfügung in einen Vektor zur Vermehrung, ohne dass die ursprüngliche DNA-Sequenz verändert wird. Nach der Isolierung können molekulare Klone verwendet werden, um viele Kopien der DNA für die Analyse der Gensequenz zu erzeugen und/oder das resultierende Protein zu exprimieren, um die Funktion des Proteins zu untersuchen oder zu nutzen. Die Klone können auch in vitro manipuliert und mutiert werden, um die Expression und Funktion des Proteins zu verändern.
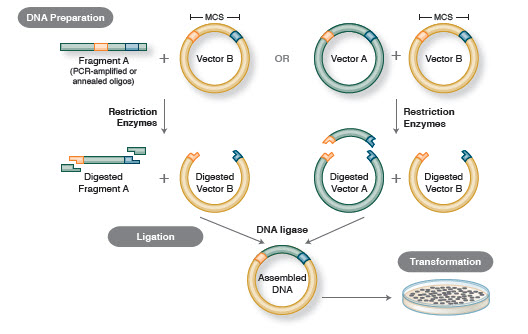
Der grundlegende Arbeitsablauf beim Klonen umfasst vier Schritte:
- Isolierung von Ziel-DNA-Fragmenten (oft als Inserts bezeichnet)
- Ligation von Inserts in einen geeigneten Klonierungsvektor, wodurch rekombinante Moleküle (z. B., Plasmide)
- Transformation rekombinanter Plasmide in Bakterien oder andere für die Vermehrung geeignete Wirte
- Screening/Auswahl von Wirten, die das gewünschte rekombinante Plasmid enthalten
Diese vier bahnbrechenden Schritte wurden in den späten 1960er und frühen 1970er Jahren von mehreren Laboratorien sorgfältig zusammengefügt und durchgeführt. Eine Zusammenfassung der Entdeckungen, die das traditionelle molekulare Klonen ausmachen, wird auf den folgenden Seiten beschrieben.
Geschichte des Klonens
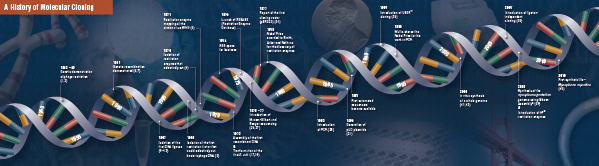
Die Grundlagen des molekularen Klonens
Schneiden (Verdauung). Die Technologie der rekombinanten DNA entstand Ende der 1960er Jahre mit der Entdeckung von Enzymen, die spezifisch doppelsträngige DNA-Moleküle schneiden und verbinden können. Bereits 1952 stellten zwei Gruppen unabhängig voneinander fest, dass Bakterien einen „Restriktionsfaktor“ kodieren, der Bakteriophagen daran hindert, in bestimmten Wirten zu wachsen (1,2). Die Natur dieses Faktors wurde jedoch erst 1968 entdeckt, als es Arber und Linn gelang, ein Enzym zu isolieren, das als Restriktionsfaktor bezeichnet wird und selektiv exogene DNA, nicht aber bakterielle DNA schneidet (3). In diesen Studien wurde auch ein Methylase-Enzym identifiziert, das die bakterielle DNA vor Restriktionsenzymen schützte.
Kurz nach der Entdeckung von Arber und Linn erweiterte und bestätigte Smith diese Studien durch die Isolierung eines Restriktionsenzyms aus Haemophilus influenza. Er wies nach, dass das Enzym selektiv DNA in der Mitte eines bestimmten 6-Basen-Paare-Abschnitts der DNA schneidet; ein Merkmal bestimmter Restriktionsenzyme ist ihre Neigung, das DNA-Substrat in oder in der Nähe von spezifischen, oft palindromischen „Erkennungs“-Sequenzen zu schneiden (4).
Die volle Leistungsfähigkeit der Restriktionsenzyme wurde erst erkannt, als Restriktionsenzyme und Gelelektrophorese zur Kartierung des Genoms des Simian Virus 40 (SV40) eingesetzt wurden (5). Für diese bahnbrechenden Erkenntnisse erhielten Werner Arber, Hamilton Smith und Daniel Nathans 1978 gemeinsam den Nobelpreis für Medizin.
Abbildung 1. Traditioneller Klonierungs-Workflow
Assembling (Ligation). Ähnlich wie der Entdeckung von Enzymen, die DNA schneiden, gingen auch der Entdeckung eines Enzyms, das DNA zusammenfügen kann, frühere, auffällige Beobachtungen voraus. Anfang der 1960er Jahre entdeckten zwei Gruppen, dass die genetische Rekombination durch das Brechen und Ligieren von DNA-Molekülen erfolgen kann (6,7), dicht gefolgt von der Beobachtung, dass lineare Bakteriophagen-DNA nach der Infektion des Wirts schnell in kovalent geschlossene Kreise umgewandelt wird (8). Nur zwei Jahre später isolierten fünf Gruppen unabhängig voneinander DNA-Ligasen und wiesen deren Fähigkeit nach, zwei DNA-Stücke zusammenzufügen (9-13).
Nicht lange nach der Entdeckung von Restriktionsenzymen und DNA-Ligasen wurde das erste rekombinante DNA-Molekül hergestellt. 1972 schnitt und ligierte Berg ein Stück Lambda-Bakteriophagen-DNA oder das Galaktose-Operon von E. coli separat mit SV40-DNA, um die ersten rekombinanten DNA-Moleküle herzustellen (14). Diese Studien waren wegweisend für das Konzept, dass aufgrund des universellen Charakters der DNS DNS aus allen Spezies miteinander verbunden werden kann. 1980 teilte sich Paul Berg den Nobelpreis für Chemie mit Walter Gilbert und Frederick Sanger (den Entwicklern der DNA-Sequenzierung) für „seine grundlegenden Studien über die Biochemie der Nukleinsäuren unter besonderer Berücksichtigung der rekombinanten DNA.“
Transformation. Die Technologie der rekombinanten DNA wäre stark eingeschränkt und das molekulare Klonen unmöglich, wenn es nicht möglich wäre, das neu konstruierte DNA-Molekül zu vermehren und zu isolieren. Die Fähigkeit, Bakterien zu transformieren oder die Aufnahme, den Einbau und die Expression fremden genetischen Materials zu induzieren, wurde erstmals von Griffith demonstriert, als er einen nicht-tödlichen Bakterienstamm in einen letalen Stamm transformierte, indem er den nicht-tödlichen Stamm mit hitzeinaktivierten letalen Bakterien mischte (15). Die Art des „Umwandlungsprinzips“, das die Letalität bewirkt, wurde jedoch bis 1944 nicht verstanden. Im selben Jahr wiesen Avery, Macleod und McCarty nach, dass nicht Proteine, sondern DNA für die Auslösung des letalen Phänotyps verantwortlich ist (16).
Anfänglich glaubte man, dass der übliche Bakterienstamm E. coli für die Transformation widerstandsfähig sei, bis Mandel und Higa nachwiesen, dass die Behandlung von E. coli mit Kalziumchlorid die Aufnahme von Bakteriophagen-DNA bewirkte (17). Cohen wandte dieses Prinzip 1972 an, als er Pionierarbeit bei der Transformation von Bakterien mit Plasmiden leistete, um den Bakterien eine Antibiotikaresistenz zu verleihen (18).
Das ultimative Experiment: Verdauung, Ligation und Transformation eines rekombinanten DNA-Moleküls wurde 1973 von Boyer, Cohen und Chang durchgeführt, als sie das Plasmid pSC101 mit EcoRI verdauten, das linearisierte Fragment mit einem anderen enzymbeschränkten Plasmid ligierten und das resultierende rekombinante Molekül in E. coli und verliehen den Bakterien eine Tetrazyklin-Resistenz (19), womit sie den Grundstein für die meisten rekombinanten DNA-Arbeiten seither legten.
Auf den Grundlagen aufbauen
Während die Wissenschaftler alle grundlegenden Prinzipien für die Erzeugung und Vermehrung rekombinanter DNA in Bakterien entdeckt und angewandt hatten, war der Prozess ineffizient. Restriktionsenzympräparate waren aufgrund nicht standardisierter Reinigungsverfahren unzuverlässig, Plasmide für die Klonierung waren umständlich, schwer zu handhaben und in ihrer Anzahl begrenzt, und die Experimente waren durch die Menge an Insert-DNA, die isoliert werden konnte, begrenzt. Die Forschung der nächsten Jahrzehnte führte zu Verbesserungen der Techniken und Werkzeuge für das molekulare Klonen.
Frühes Vektordesign.
Entwicklung des ersten standardisierten Vektors. Wissenschaftler, die in Boyers Labor arbeiteten, erkannten den Bedarf an einem allgemeinen Klonierungsplasmid, einem kompakten Plasmid mit eindeutigen Restriktionsstellen für die Klonierung von Fremd-DNA und die Expression von Antibiotikaresistenzgenen für die Selektion von transformierten Bakterien. Im Jahr 1977 beschrieben sie den ersten Vektor für Klonierungszwecke, pBR322 (20). Dieser Vektor war klein, ~4 Kilobasen groß, und enthielt zwei Antibiotikaresistenzgene für die Selektion.
Vektoren mit On-Board-Screening und höheren Ausbeuten. Obwohl die Antibiotikaselektion das Wachstum nicht-transformierter Bakterien verhinderte, konnten Plasmide, die ohne Insert-DNA-Fragmente re-ligierten (Selbstligation), den Bakterien immer noch eine Antibiotikaresistenz verleihen. Daher konnte es zeitaufwändig sein, die richtigen Bakterienklone zu finden, die das gewünschte rekombinante DNA-Molekül enthielten.
Vieira und Messing entwickelten ein Screening-Tool, um Bakterienkolonien zu identifizieren, die Plasmide mit DNA-Insertionen enthielten. Auf der Grundlage des Plasmids pBR322 schufen sie die Serie der pUC-Plasmide, die ein „blau/weißes Screening“-System enthielten (21). Durch die Platzierung einer multiplen Klonierungsstelle (MCS) mit mehreren einzigartigen Restriktionsstellen innerhalb des LacZ‘-Gens konnten die Forscher nach Bakterienkolonien suchen, die Plasmide mit dem fremden DNA-Insert enthielten. Wenn Bakterien auf den richtigen Medien plattiert wurden, enthielten weiße Kolonien Plasmide mit Inserts, während blaue Kolonien Plasmide ohne Inserts enthielten. pUC-Plasmide hatten einen zusätzlichen Vorteil gegenüber bestehenden Vektoren: Sie enthielten eine Mutation, die zu einer höheren Kopienzahl führte und somit die Plasmidausbeute erhöhte.
Verbesserung von Restriktionsverdauungen. Die frühe Arbeit mit Restriktionsenzymen wurde durch die Reinheit der Enzympräparate und die mangelnde Kenntnis der Pufferanforderungen für die einzelnen Enzyme behindert. Im Jahr 1975 brachte New England Biolabs (NEB) als erstes Unternehmen Restriktionsenzyme auf den Markt, die aus einer rekombinanten Quelle hergestellt wurden. Dies ermöglichte eine höhere Ausbeute, eine bessere Reinheit, eine gleichbleibende Qualität von Charge zu Charge und niedrigere Preise. Derzeit wurden von Wissenschaftlern auf der ganzen Welt über 4.000 Restriktionsenzyme entdeckt, die über 300 verschiedene Sequenzen erkennen. NEB bietet derzeit über 230 dieser Spezifitäten an.
NEB war auch eines der ersten Unternehmen, das ein standardisiertes Vier-Puffer-System entwickelte und alle seine Enzymaktivitäten in diesem Puffersystem charakterisierte. Dies führte zu einem besseren Verständnis der Durchführung eines Doppelverdaus, d. h. des Verdaus der DNA mit zwei Enzymen gleichzeitig. Spätere Forschungen führten zur Entwicklung von Ein-Puffer-Systemen, die mit den gebräuchlichsten Restriktionsenzymen kompatibel sind (z. B. CutSmart™ Buffer von NEB).
Mit dem Aufkommen kommerziell erhältlicher Restriktionsenzymbibliotheken mit bekannten Sequenzspezifitäten wurden Restriktionsenzyme zu einem leistungsfähigen Werkzeug für das Screening potenzieller rekombinanter DNA-Klone. Der „diagnostische Verdau“ war und ist immer noch eine der am häufigsten verwendeten Techniken bei der molekularen Klonierung.
Vektor- und Insertpräparation. Die Effizienz und Vielseitigkeit der Klonierung wurde auch durch die Entwicklung verschiedener Techniken zur Vorbereitung der Vektoren vor der Ligation verbessert. Es wurden alkalische Phosphatasen isoliert, die die 3′- und 5′-Phosphatgruppen von den Enden der DNA entfernen konnten. Bald entdeckte man, dass die Behandlung von Vektoren mit Kälberdarm-Phosphatase (CIP) DNA-Enden dephosphoryliert und die Selbstligierung des Vektors verhindert, wodurch die Wiedergewinnung von Plasmiden mit Insert (24) erhöht wird.
Das CIP-Enzym erwies sich als schwierig zu inaktivieren, und jede Restaktivität führte zur Dephosphorylierung der Insert-DNA und zur Hemmung der Ligationsreaktion. Die Entdeckung der hitzelabilen alkalischen Phosphatasen, wie die rekombinante alkalische Phosphatase der Garnele (rSAP) und die antarktische Phosphatase (AP) (beide von NEB vertrieben), verringerte die erforderlichen Schritte und den Zeitaufwand, da eine einfache Temperaturveränderung das Enzym vor dem Ligationsschritt inaktiviert (25).
Die DNA-Sequenzierung kommt an. Die DNA-Sequenzierung wurde in den späten 1970er Jahren entwickelt, als zwei konkurrierende Methoden entwickelt wurden. Maxam und Gilbert entwickelten die „chemische Sequenzierungsmethode“, die auf der chemischen Modifizierung der DNA und der anschließenden Spaltung an bestimmten Basen beruhte (26). Zur gleichen Zeit veröffentlichten Sanger und Kollegen die „Kettenabbruchmethode“, die von den meisten Forschern verwendet wurde (27). Die Sanger-Methode wurde schnell automatisiert, und die ersten automatischen Sequenziergeräte wurden 1987 verkauft.
Die Möglichkeit, die Sequenz eines DNA-Abschnitts zu bestimmen, erhöhte die Zuverlässigkeit und Vielseitigkeit des molekularen Klonens. Nach dem Klonen konnten die Wissenschaftler die Klone sequenzieren, um das richtige rekombinante Molekül definitiv zu identifizieren, neue Gene oder Mutationen in Genen zu erkennen und auf einfache Weise Oligonukleotide auf der Grundlage der bekannten Sequenz für weitere Experimente zu entwickeln.
Die Auswirkungen der Polymerase-Kettenreaktion. Eines der Probleme bei der molekularen Klonierung in den Anfangsjahren bestand darin, genügend Insert-DNA zu erhalten, um sie in den Vektor zu klonen. Im Jahr 1983 entwickelte Mullis eine Technik, die dieses Problem löste und das molekulare Klonen revolutionierte (28). Er amplifizierte einen Abschnitt der Ziel-DNA, indem er entgegengesetzte Primer verwendete, um beide komplementären DNA-Stränge gleichzeitig zu amplifizieren. Durch Denaturierungs-, Annealing- und Polymerisationszyklen zeigte er, dass er eine einzelne DNA-Kopie exponentiell vervielfältigen konnte. Die Polymerase-Kettenreaktion (PCR) ermöglichte die Vervielfältigung und das Klonen von Genen aus zuvor unzureichenden DNA-Mengen. Für diese Entdeckung erhielt Kary Mullis 1993 den Nobelpreis für Chemie „für Beiträge zur Entwicklung von Methoden in der DNA-basierten Chemie“.
Im Jahr 1970 entdeckten Temin und Baltimore unabhängig voneinander die reverse Transkriptase in Viren, ein Enzym, das RNA in DNA umwandelt (29,30). Kurz nach der Entwicklung der PCR wurde die reverse Transkription mit der PCR (RT-PCR) gekoppelt, um das Klonen von Boten-RNA (mRNA) zu ermöglichen. Mit Hilfe der reversen Transkription wurde eine DNA-Kopie (cDNA) der mRNA erstellt, die anschließend durch PCR amplifiziert wurde, um ein Insert für die Ligation zu erzeugen. Für ihre Entdeckung des Enzyms erhielten Howard Temin und David Baltimore 1975 den Nobelpreis für Medizin und Physiologie, den sie sich mit Renato Dulbecco teilten.
Klonierung von PCR-Produkten. Das Aufkommen der PCR bedeutete, dass Forscher nun Gene und DNA-Abschnitte mit begrenztem Wissen über die Amplikonsequenz klonen konnten. Es gab jedoch kaum einen Konsens über die optimale Methode zur Vorbereitung von PCR-Produkten für eine effiziente Ligation in Klonierungsvektoren.
Zunächst wurden verschiedene Methoden zur Klonierung von PCR-Produkten verwendet. Die einfachste und immer noch gebräuchlichste Methode zur Klonierung von PCR-Produkten ist die Einführung von Restriktionsstellen an den Enden des PCR-Produkts (31). Dies ermöglicht eine direkte, gerichtete Klonierung des Inserts in den Vektor nach dem Restriktionsverdau. Die Klonierung mit stumpfen Enden wurde entwickelt, um PCR-Produkte direkt zu ligieren, die von Polymerasen erzeugt wurden, die stumpfe Enden produzieren, oder Inserts, die so konstruiert wurden, dass sie Restriktionsstellen aufweisen, die stumpfe Enden hinterlassen, sobald das Insert verdaut wurde. Dies war nützlich bei der Klonierung von DNA-Fragmenten, die keine mit dem Vektor kompatiblen Restriktionsstellen enthielten (32).
Kurz nach der Einführung der PCR wurde die Overlap-Extension-PCR als Methode zum Zusammenfügen von PCR-Produkten zu einer einzigen zusammenhängenden DNA-Sequenz eingeführt (33). Bei dieser Methode wird das DNA-Insert durch PCR mit Primern amplifiziert, die ein PCR-Produkt erzeugen, das mit dem Vektor überlappende Regionen enthält. Der Vektor und das Insert werden dann gemischt, denaturiert und annealed, was eine Hybridisierung des Inserts mit dem Vektor ermöglicht. In einer zweiten PCR-Runde werden rekombinante DNA-Moleküle des inserthaltigen Vektors erzeugt. Die Overlap-Extension-PCR ermöglichte es den Forschern, große Gene zusammenzusetzen, die mit herkömmlichen PCR-Methoden nicht ohne weiteres amplifiziert werden konnten. Die Overlap-Extension-PCR wurde auch verwendet, um Mutationen in Gensequenzen einzuführen (34).
Abbildung 2. Überblick über die PCR

Entwicklung spezialisierter Klonierungstechniken.
In dem Bestreben, die Effizienz der molekularen Klonierung weiter zu verbessern, wurden mehrere spezialisierte Werkzeuge und Techniken entwickelt, die die Eigenschaften einzigartiger Enzyme nutzen.
TA-Klonierung. Ein Ansatz nutzte eine Eigenschaft der Taq-DNA-Polymerase, der ersten hitzestabilen Polymerase, die für die PCR verwendet wurde. Während der Amplifikation fügt die Taq ein einzelnes 3′-DA-Nukleotid an das Ende jedes PCR-Produkts an. Das PCR-Produkt kann leicht in einen Vektor ligiert werden, der so geschnitten und konstruiert wurde, dass er einzelne T-Reste auf jedem Strang enthält. Mehrere Unternehmen haben diese Technik vermarktet und verkaufen Kits, die bereits linearisierte und „geschwänzte“ Klonierungsvektoren enthalten.
LIC. Die ligationsunabhängige Klonierung (LIC) ermöglicht, wie der Name schon sagt, die Verbindung von DNA-Molekülen in Abwesenheit von DNA-Ligase. LIC wird üblicherweise mit T4-DNA-Polymerase durchgeführt, die zur Erzeugung von einzelsträngigen DNA-Überhängen von >12 Nukleotiden Länge sowohl auf der linearisierten Vektor-DNA als auch auf dem zu klonierenden Insert verwendet wird (35). Wenn sie zusammengemischt werden, verbinden sich der Vektor und das Insert durch die lange Strecke der kompatiblen Enden. Die Länge der kompatiblen Enden reicht aus, um das Molekül in Abwesenheit von Ligase zusammenzuhalten, selbst während der Transformation. Nach der Transformation werden die Lücken in vivo repariert. Im Handel sind verschiedene Produkte für das LIC.USER-Klonen erhältlich.
Das USER-Klonen wurde in den frühen 1990er Jahren als Restriktionsenzym- und Ligase-unabhängige Klonierungsmethode entwickelt (36). Bei der Entwicklung der Methode wurden PCR-Primer verwendet, die einen 5′-Schwanz von etwa 12 Nukleotiden enthielten, in dem mindestens vier Desoxythymidinbasen durch Desoxyuridine ersetzt worden waren. Das PCR-Produkt wurde mit Uracil-DNA-Glykosidase (UDG) und Endonuklease VIII behandelt, wodurch die Uracil-Basen entfernt wurden und eine 3′-Überlappung übrig blieb, die an einen ähnlich behandelten Vektor angehängt werden konnte. NEB vertreibt das USER-Enzym für Ligase- und Restriktionsenzym-unabhängige Klonierungsreaktionen.
Zukunftstrends
Die molekulare Klonierung hat sich von der Klonierung eines einzelnen DNA-Fragments zur Zusammenfügung mehrerer DNA-Komponenten zu einem einzigen zusammenhängenden DNA-Abschnitt entwickelt. Neue und aufkommende Technologien versuchen, das Klonen in einen Prozess zu verwandeln, der so einfach ist wie das Anordnen von DNA-Blöcken nebeneinander.
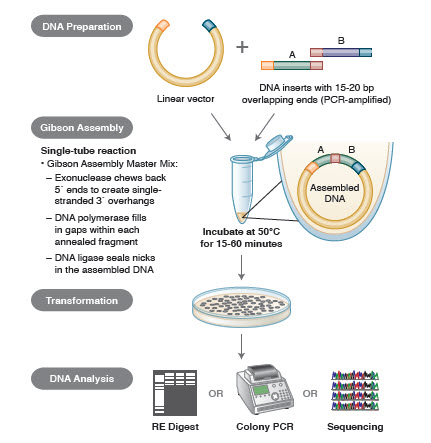
DNA-Assembly-Methoden. Viele neue, elegante Technologien ermöglichen den Zusammenbau mehrerer DNA-Fragmente in einer Ein-Rohr-Reaktion. Die Vorteile dieser Technologien sind, dass sie standardisiert, nahtlos und weitgehend sequenzunabhängig sind. Darüber hinaus wird durch die Möglichkeit, mehrere DNA-Fragmente in einem Röhrchen zu assemblieren, eine Reihe von zuvor unabhängigen Restriktions-/Ligationsreaktionen zu einem rationalisierten, effizienten Verfahren.
Zu den verschiedenen Techniken und Produkten für die Genassemblierung gehören SLIC (Sequence and Ligase Independent Cloning), Gibson Assembly (NEB), GeneArt® Seamless Cloning (Life Technologies) und Gateway® Cloning (Invitrogen) (35,37,38).
Bei der DNA-Assemblierung werden die zu assemblierenden DNA-Blöcke mittels PCR amplifiziert. Dann werden die DNA-Fragmente, die nebeneinander montiert werden sollen, so bearbeitet, dass sie Blöcke komplementärer Sequenzen enthalten, die miteinander ligiert werden. Dabei kann es sich um kompatible kohäsive Enden handeln, wie sie bei der Gibson Assembly verwendet werden, oder um Regionen, die Erkennungsstellen für ortsspezifische Rekombinasen (Gateway) enthalten. Das für die DNA-Ligation verwendete Enzym erkennt jeden Satz kompatibler Regionen und fügt sie zusammen, so dass in einer Reaktion ein einziges, zusammenhängendes DNA-Molekül entsteht.
Abbildung 3. Überblick über die Gibson Assembly Klonierungsmethode
Synthetische Biologie. Die DNA-Synthese ist ein Bereich der synthetischen Biologie, der derzeit die rekombinante DNA-Technologie revolutioniert. Obwohl 1972 erstmals ein komplettes Gen in vitro synthetisiert wurde (40), wurde die DNA-Synthese großer DNA-Moleküle erst in den frühen 2000er Jahren Realität, als Forscher begannen, ganze Genome in vitro zu synthetisieren (41,42). Diese frühen Experimente dauerten Jahre, aber die Technologie beschleunigt die Fähigkeit, große DNA-Moleküle zu synthetisieren.
Schlussfolgerung
In den letzten 40 Jahren hat sich das molekulare Klonen weiterentwickelt, von der mühsamen Isolierung und dem Zusammenfügen von zwei DNA-Stücken, gefolgt von einem intensiven Screening potenzieller Klone, bis hin zum nahtlosen Zusammenfügen von bis zu zehn DNA-Fragmenten mit bemerkenswerter Effizienz in nur wenigen Stunden, oder dem Design von DNA-Molekülen in silico und ihrer Synthese in vitro. Alle diese Technologien zusammen geben den Molekularbiologen ein erstaunlich leistungsfähiges Instrumentarium zur Erforschung, Manipulation und Nutzung der DNA an die Hand, das die Horizonte der Wissenschaft weiter erweitern wird. Zu den Möglichkeiten gehören die Entwicklung sicherer rekombinanter Proteine für die Behandlung von Krankheiten, die Verbesserung der Gentherapie (43) und die schnellere Herstellung, Validierung und Freigabe neuer Impfstoffe (44). Aber letztlich wird das Potenzial nur durch unsere Vorstellungskraft begrenzt.
Rebecca Tirabassi ist Redaktionsassistentin bei Bitesizebio.com.