La clonación molecular, término que ha llegado a significar la creación de moléculas de ADN recombinante, ha impulsado el progreso en todas las ciencias de la vida. A partir de la década de 1970, con el descubrimiento de las endonucleasas de restricción (enzimas que cortan moléculas de ADN de forma selectiva y específica), la tecnología del ADN recombinante ha experimentado un crecimiento exponencial tanto en su aplicación como en su sofisticación, dando lugar a herramientas cada vez más potentes para la manipulación del ADN. La clonación de genes es ahora tan sencilla y eficaz que se ha convertido en una técnica estándar de laboratorio. Esto ha llevado a una explosión en la comprensión de la función de los genes en las últimas décadas. Las tecnologías emergentes prometen posibilidades aún mayores, como permitir a los investigadores unir sin problemas múltiples fragmentos de ADN y transformar los plásmidos resultantes en bacterias, en menos de dos horas, o el uso de casetes de genes intercambiables, que pueden moverse fácilmente entre diferentes construcciones, para maximizar la velocidad y la flexibilidad. En un futuro próximo, la clonación molecular verá probablemente la aparición de un nuevo paradigma, con técnicas de biología sintética que permitirán la síntesis química in vitro de cualquier construcción de ADN especificada in silico. Estos avances deberían permitir una construcción e iteración más rápida de los clones de ADN, acelerando el desarrollo de vectores de terapia génica, procesos de producción de proteínas recombinantes y nuevas vacunas.
Rebecca Tirabassi, Bitesize Bio.
- Introducción
- Historia de la clonación
- La base de la clonación molecular
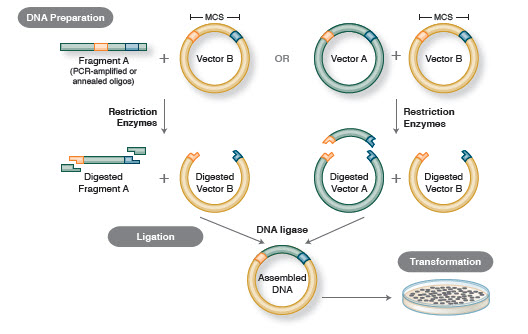
- Figura 1. Flujo de trabajo de clonación tradicional
- Construcción de la base
- Figura 2. Visión general de la PCR
- Tendencias futuras
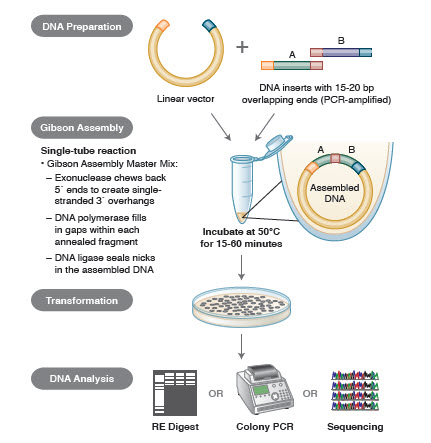
- Figura 3. Visión general del método de clonación por ensamblaje Gibson
- Conclusión
- Vea nuestra Guía Técnica de Clonación Molecular
Introducción
La clonación molecular se refiere al aislamiento de una secuencia de ADN de cualquier especie (a menudo un gen), y su inserción en un vector para su propagación, sin alterar la secuencia de ADN original. Una vez aislados, los clones moleculares pueden utilizarse para generar muchas copias del ADN para el análisis de la secuencia del gen, y/o para expresar la proteína resultante para el estudio o la utilización de la función de la proteína. Los clones también pueden manipularse y mutarse in vitro para alterar la expresión y la función de la proteína.
El flujo de trabajo básico de la clonación incluye cuatro pasos:
- Aislamiento de los fragmentos de ADN objetivo (a menudo denominados insertos)
- Ligación de los insertos en un vector de clonación apropiado, creando moléculas recombinantes (por ejemplo, plásmidos)
- Transformación de plásmidos recombinantes en bacterias u otro huésped adecuado para su propagación
- Cribado/selección de huéspedes que contengan el plásmido recombinante deseado
Estos cuatro pasos pioneros fueron cuidadosamente unidos y llevados a cabo por múltiples laboratorios, a partir de finales de los años 60 y principios de los 70. En las siguientes páginas se describe un resumen de los descubrimientos que conforman la clonación molecular tradicional.
Historia de la clonación
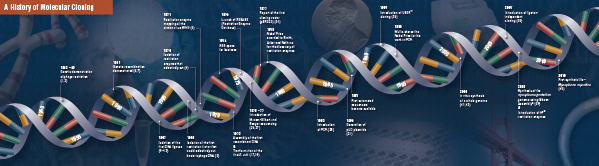
La base de la clonación molecular
Corte (digestión). La tecnología del ADN recombinante surgió por primera vez a finales de la década de 1960, con el descubrimiento de enzimas que podían cortar y unir específicamente moléculas de ADN de doble cadena. De hecho, ya en 1952, dos grupos observaron de forma independiente que las bacterias codificaban un «factor de restricción» que impedía que los bacteriófagos crecieran dentro de ciertos huéspedes (1,2). Pero la naturaleza del factor no se descubrió hasta 1968, cuando Arber y Linn consiguieron aislar una enzima, denominada factor de restricción, que cortaba selectivamente el ADN exógeno, pero no el bacteriano (3). Estos estudios también identificaron una enzima metilasa que protegía el ADN bacteriano de las enzimas de restricción.
Poco después del descubrimiento de Arber y Linn, Smith amplió y confirmó estos estudios aislando una enzima de restricción de Haemophilus influenza. Demostró que la enzima cortaba selectivamente el ADN en el centro de un tramo específico de 6 pares de bases de ADN; una característica de ciertas enzimas de restricción es su propensión a cortar el sustrato de ADN en o cerca de secuencias de «reconocimiento» específicas, a menudo palindrómicas (4).
No se comprendió todo el poder de las enzimas de restricción hasta que se utilizaron las enzimas de restricción y la electroforesis en gel para cartografiar el genoma del virus simio 40 (SV40) (5). Por estos hallazgos fundamentales, Werner Arber, Hamilton Smith y Daniel Nathans compartieron el Premio Nobel de Medicina de 1978.
Figura 1. Flujo de trabajo de clonación tradicional
Assembling (Ligation). Al igual que el descubrimiento de las enzimas que cortan el ADN, el descubrimiento de una enzima que podía unir el ADN fue precedido por observaciones anteriores y destacadas. A principios de la década de 1960, dos grupos descubrieron que la recombinación genética podía producirse a través de la rotura y la ligadura de moléculas de ADN (6,7), seguida de cerca por la observación de que el ADN lineal de los bacteriófagos se convierte rápidamente en círculos cerrados covalentemente tras la infección del huésped (8). Apenas dos años más tarde, cinco grupos aislaron de forma independiente las ADN ligasas y demostraron su capacidad para ensamblar dos trozos de ADN (9-13).
No mucho después del descubrimiento de las enzimas de restricción y las ADN ligasas, se fabricó la primera molécula de ADN recombinante. En 1972, Berg cortó y ligó por separado un trozo de ADN del bacteriófago lambda o del operón de galactosa de E. coli con ADN SV40 para crear las primeras moléculas de ADN recombinante (14). Estos estudios fueron pioneros en el concepto de que, debido a la naturaleza universal del ADN, se podía unir ADN de cualquier especie. En 1980, Paul Berg compartió el Premio Nobel de Química con Walter Gilbert y Frederick Sanger (los desarrolladores de la secuenciación del ADN), por «sus estudios fundamentales de la bioquímica de los ácidos nucleicos, con especial atención al ADN recombinante.»
Transformación. La tecnología del ADN recombinante estaría muy limitada, y la clonación molecular sería imposible, sin los medios para propagar y aislar la molécula de ADN recién construida. La capacidad de transformar bacterias, o de inducir la captación, incorporación y expresión de material genético extraño, fue demostrada por primera vez por Griffith cuando transformó una cepa no letal de bacterias en una cepa letal mezclando la cepa no letal con bacterias letales inactivadas por calor (15). Sin embargo, la naturaleza del «principio transformador» que transmitía la letalidad no se comprendió hasta 1944. Ese mismo año, Avery, Macleod y McCarty demostraron que era el ADN, y no las proteínas, el responsable de inducir el fenotipo letal (16).
Al principio se creía que la cepa bacteriana común de laboratorio, E. coli, era refractaria a la transformación, hasta que Mandel y Higa demostraron que el tratamiento de E. coli con cloruro de calcio inducía la captación de ADN bacteriófago (17). Cohen aplicó este principio, en 1972, cuando fue pionero en la transformación de bacterias con plásmidos para conferirles resistencia a los antibióticos (18).
El experimento definitivo: digestión, ligadura y transformación de una molécula de ADN recombinante fue ejecutado por Boyer, Cohen y Chang en 1973, cuando digirieron el plásmido pSC101 con EcoRI, ligaron el fragmento linealizado a otro plásmido de enzima restringida y transformaron la molécula recombinante resultante en E. coli, confiriendo a la bacteria resistencia a la tetraciclina (19), sentando así las bases de la mayor parte de los trabajos sobre ADN recombinante realizados desde entonces.
Construcción de la base
Aunque los científicos habían descubierto y aplicado todos los principios básicos para crear y propagar el ADN recombinante en bacterias, el proceso era ineficiente. Las preparaciones de enzimas de restricción eran poco fiables debido a los procedimientos de purificación no estandarizados, los plásmidos para la clonación eran engorrosos, difíciles de trabajar y limitados en número, y los experimentos estaban limitados por la cantidad de ADN de inserción que se podía aislar. Las investigaciones de las décadas siguientes permitieron mejorar las técnicas y herramientas disponibles para la clonación molecular.
Diseño temprano de vectores.
Desarrollo del primer vector estandarizado. Los científicos que trabajaban en el laboratorio de Boyer reconocieron la necesidad de un plásmido de clonación general, un plásmido compacto con sitios de restricción únicos para la clonación en ADN extraño y la expresión de genes de resistencia a los antibióticos para la selección de bacterias transformadas. En 1977, describieron el primer vector diseñado con fines de clonación, el pBR322 (20). Este vector era pequeño, con un tamaño de ~4 kilobases, y tenía dos genes de resistencia a los antibióticos para su selección.
Vectores con selección incorporada y mayores rendimientos. Aunque la selección de antibióticos impedía el crecimiento de las bacterias no transformadas, los plásmidos que se religaban sin fragmentos de ADN de inserción (autoligado) podían seguir confiriendo resistencia a los antibióticos a las bacterias. Por lo tanto, encontrar los clones bacterianos correctos que contenían la molécula de ADN recombinante deseada podía llevar mucho tiempo.
Vieira y Messing idearon una herramienta de cribado para identificar las colonias bacterianas que contenían plásmidos con inserciones de ADN. Basándose en el plásmido pBR322, crearon la serie de plásmidos pUC, que contenían un sistema de «cribado azul/blanco» (21). La colocación de un sitio de clonación múltiple (MCS) que contenía varios sitios de restricción únicos dentro del gen LacZ’ permitió a los investigadores realizar un cribado de las colonias bacterianas que contenían plásmidos con el inserto de ADN extraño. Los plásmidos pUC tenían una ventaja adicional sobre los vectores existentes: contenían una mutación que daba lugar a un mayor número de copias, lo que aumentaba el rendimiento de los plásmidos.
Mejora de las digestiones de restricción. Los primeros trabajos con enzimas de restricción se vieron obstaculizados por la pureza de la preparación de las enzimas y la falta de conocimiento de los requisitos de los tampones para cada enzima. En 1975, New England Biolabs (NEB) se convirtió en la primera empresa en comercializar enzimas de restricción producidas a partir de una fuente recombinante. Esto permitió un mayor rendimiento, una mayor pureza, una consistencia entre lotes y un precio más bajo. Actualmente, los científicos de todo el mundo han descubierto más de 4.000 enzimas de restricción que reconocen más de 300 secuencias diferentes. NEB suministra actualmente más de 230 de estas especificidades.
NEB fue también una de las primeras empresas en desarrollar un sistema estandarizado de cuatro tampones, y en caracterizar todas sus actividades enzimáticas en este sistema de tampones. Esto condujo a una mejor comprensión de cómo llevar a cabo una doble digestión, o la digestión del ADN con dos enzimas simultáneamente. La investigación posterior condujo al desarrollo de sistemas de tampón único, que son compatibles con las enzimas de restricción más comunes (como el tampón CutSmart™ de NEB).
Con la llegada de las bibliotecas de enzimas de restricción disponibles en el mercado con especificidades de secuencia conocidas, las enzimas de restricción se convirtieron en una poderosa herramienta para el cribado de posibles clones de ADN recombinante. La «digestión de diagnóstico» fue, y sigue siendo, una de las técnicas más comunes utilizadas en la clonación molecular.
Preparación de vectores e insertos. La eficacia y la versatilidad de la clonación también mejoraron con el desarrollo de diferentes técnicas para preparar los vectores antes de la ligadura. Se aislaron fosfatasas alcalinas que podían eliminar los grupos fosfato 3′ y 5′ de los extremos del ADN . Pronto se descubrió que el tratamiento de los vectores con fosfatasa intestinal de ternera (CIP) desfosforilaba los extremos del ADN e impedía la autoligación del vector, lo que aumentaba la recuperación de los plásmidos con el inserto (24).
La enzima CIP resultó difícil de inactivar, y cualquier actividad residual provocaba la desfosforilación del ADN del inserto y la inhibición de la reacción de ligación. El descubrimiento de las fosfatasas alcalinas termolábiles, como la fosfatasa alcalina de camarón recombinante (rSAP) y la fosfatasa antártica (AP) (ambas vendidas por NEB), disminuyó los pasos y el tiempo necesario, ya que un simple cambio de temperatura inactiva la enzima antes del paso de ligación (25).
Llega la secuenciación del ADN. La secuenciación del ADN se desarrolló a finales de la década de 1970, cuando se idearon dos métodos que competían entre sí. Maxam y Gilbert desarrollaron el «método de secuenciación química», que se basaba en la modificación química del ADN y la posterior escisión en bases específicas (26). Al mismo tiempo, Sanger y sus colegas publicaron el «método de terminación en cadena», que se convirtió en el método utilizado por la mayoría de los investigadores (27). El método de Sanger se automatizó rápidamente y los primeros secuenciadores automáticos se vendieron en 1987.
La capacidad de determinar la secuencia de un tramo de ADN mejoró la fiabilidad y versatilidad de la clonación molecular. Una vez clonados, los científicos podían secuenciar los clones para identificar definitivamente la molécula recombinante correcta, identificar nuevos genes o mutaciones en los genes y diseñar fácilmente oligonucleótidos basados en la secuencia conocida para experimentos adicionales.
El impacto de la reacción en cadena de la polimerasa. Uno de los problemas de la clonación molecular en los primeros años era obtener suficiente ADN de inserción para clonar en el vector. En 1983, Mullis ideó una técnica que resolvió este problema y revolucionó la clonación molecular (28). Amplificó un tramo de ADN objetivo utilizando cebadores opuestos para amplificar ambas cadenas complementarias de ADN, simultáneamente. Mediante ciclos de desnaturalización, recocido y polimerización, demostró que podía amplificar exponencialmente una sola copia de ADN. La reacción en cadena de la polimerasa, o PCR, permitió amplificar y clonar genes a partir de cantidades de ADN hasta entonces inadecuadas. Por este descubrimiento, Kary Mullis compartió el Premio Nobel de Química de 1993 «por sus contribuciones al desarrollo de métodos dentro de la química basada en el ADN».
En 1970, Temin y Baltimore descubrieron de forma independiente la transcriptasa inversa en los virus, una enzima que convierte el ARN en ADN (29,30). Poco después de que se desarrollara la PCR, la transcripción inversa se acopló a la PCR (RT-PCR) para permitir la clonación del ARN mensajero (ARNm). La transcripción inversa se utilizó para crear una copia de ADN (ADNc) del ARNm que posteriormente se amplificó por PCR para crear un inserto para la ligadura. Por su descubrimiento de la enzima, Howard Temin y David Baltimore recibieron el Premio Nobel de Medicina y Fisiología de 1975, que compartieron con Renato Dulbecco.
Clonación de productos de PCR. La aparición de la PCR permitió a los investigadores clonar genes y segmentos de ADN con un conocimiento limitado de la secuencia del amplicón. Sin embargo, había poco consenso en cuanto al método óptimo de preparación de productos de PCR para una ligadura eficiente en vectores de clonación.
Inicialmente se utilizaron varios métodos diferentes para clonar productos de PCR. El método más sencillo, y todavía el más común, para clonar productos de PCR es la introducción de sitios de restricción en los extremos del producto de PCR (31). Esto permite la clonación directa y direccional del inserto en el vector tras la digestión de restricción. La clonación con extremos romos se desarrolló para ligar directamente productos de PCR generados por polimerasas que producían extremos romos, o insertos diseñados para tener sitios de restricción que dejaban extremos romos una vez que el inserto era digerido. Esto fue útil para clonar fragmentos de ADN que no contenían sitios de restricción compatibles con el vector (32).
Poco después de la introducción de la PCR, se introdujo la PCR de extensión solapada como método para ensamblar productos de PCR en una secuencia de ADN contigua (33). En este método, el inserto de ADN se amplifica por PCR utilizando cebadores que generan un producto de PCR que contiene regiones superpuestas con el vector. A continuación, el vector y el inserto se mezclan, se desnaturalizan y se recombinan, lo que permite la hibridación del inserto con el vector. Una segunda ronda de PCR genera moléculas de ADN recombinante del vector que contiene el inserto. La PCR de extensión de solapamiento permitió a los investigadores unir grandes genes que no podían amplificarse fácilmente con los métodos tradicionales de PCR. La PCR de extensión de solapamiento también se utilizó para introducir mutaciones en secuencias de genes (34).
Figura 2. Visión general de la PCR

Desarrollo de técnicas de clonación especializadas.
En un esfuerzo por mejorar aún más la eficiencia de la clonación molecular, se desarrollaron varias herramientas y técnicas especializadas que explotaban las propiedades de enzimas únicas.
Clonación de AT. Un enfoque aprovechó una propiedad de la ADN polimerasa Taq, la primera polimerasa termoestable utilizada para la PCR. Durante la amplificación, Taq añade un único nucleótido 3′ dA al final de cada producto de la PCR. El producto de la PCR puede ligarse fácilmente a un vector que ha sido cortado y diseñado para contener residuos T individuales en cada hebra. Varias empresas han comercializado la técnica y venden kits que contienen vectores de clonación ya linealizados y «con cola».
LIC. La clonación independiente de la ligadura (LIC), como su nombre indica, permite la unión de moléculas de ADN en ausencia de ADN ligasa. El LIC se realiza habitualmente con la ADN polimerasa T4, que se utiliza para generar salientes de ADN monocatenario, de >12 nucleótidos de longitud, tanto en el ADN del vector linealizado como en el inserto a clonar (35). Cuando se mezclan, el vector y el inserto se anejan a través del largo tramo de extremos compatibles. La longitud de los extremos compatibles es suficiente para mantener la molécula unida en ausencia de ligasa, incluso durante la transformación. Una vez transformados, los huecos se reparan in vivo. Hay varios productos diferentes disponibles en el mercado para la clonación LIC.USER.
La clonación USER se desarrolló por primera vez a principios de la década de 1990 como un método de clonación independiente de la enzima de restricción y de la ligasa (36). Cuando se concibió por primera vez, el método se basaba en el uso de cebadores de PCR que contenían una cola 5′ de ~12 nucleótidos, en la que al menos cuatro bases de desoximidina habían sido sustituidas por desoxiuridinas. El producto de la PCR se trataba con uracilo ADN glicosidasa (UDG) y Endonucleasa VIII, que extirpa las bases de uracilo y deja un solapamiento 3′ que puede ser recocido en un vector tratado de forma similar. NEB vende la enzima USER para reacciones de clonación independientes de la ligasa y la enzima de restricción.
Tendencias futuras
La clonación molecular ha progresado desde la clonación de un único fragmento de ADN hasta el ensamblaje de múltiples componentes de ADN en un único tramo contiguo de ADN. Las tecnologías nuevas y emergentes pretenden transformar la clonación en un proceso tan sencillo como disponer «bloques» de ADN uno al lado del otro.
Métodos de ensamblaje del ADN. Muchas tecnologías nuevas y elegantes permiten el ensamblaje de múltiples fragmentos de ADN en una reacción de un solo tubo. Las ventajas de estas tecnologías son que son estandarizadas, sin fisuras y en su mayoría independientes de la secuencia. Además, la capacidad de ensamblar múltiples fragmentos de ADN en un tubo convierte una serie de reacciones de restricción/ligación previamente independientes en un procedimiento racionalizado y eficiente.
Diferentes técnicas y productos para el ensamblaje de genes incluyen SLIC (Sequence and Ligase Independent Cloning), Gibson Assembly (NEB), GeneArt® Seamless Cloning (Life Technologies) y Gateway® Cloning (Invitrogen) (35,37,38).
En el ensamblaje de ADN, los bloques de ADN a ensamblar se amplifican por PCR. A continuación, los fragmentos de ADN que se van a ensamblar adyacentes entre sí se diseñan para que contengan bloques de secuencias complementarias que se ligarán entre sí. Puede tratarse de extremos cohesivos compatibles, como los utilizados para el ensamblaje Gibson, o de regiones que contengan sitios de reconocimiento para recombinasas de sitio específico (Gateway). La enzima utilizada para la ligación del ADN reconocerá y ensamblará cada conjunto de regiones compatibles, creando una única molécula de ADN contigua en una sola reacción.
Figura 3. Visión general del método de clonación por ensamblaje Gibson
Biología sintética. La síntesis de ADN es un área de la biología sintética que actualmente está revolucionando la tecnología del ADN recombinante. Aunque en 1972 se sintetizó por primera vez un gen completo in vitro (40), la síntesis de grandes moléculas de ADN no se hizo realidad hasta principios de la década de 2000, cuando los investigadores comenzaron a sintetizar genomas completos in vitro (41,42). Estos primeros experimentos tardaron años en completarse, pero la tecnología está acelerando la capacidad de sintetizar grandes moléculas de ADN.
Conclusión
En los últimos 40 años, la clonación molecular ha pasado de aislar y juntar arduamente dos trozos de ADN, seguido de un cribado intensivo de clones potenciales, a ensamblar sin problemas hasta 10 fragmentos de ADN con una eficiencia notable en sólo unas horas, o a diseñar moléculas de ADN in silico y sintetizarlas in vitro. En conjunto, todas estas tecnologías proporcionan a los biólogos moleculares una caja de herramientas asombrosamente potente para explorar, manipular y aprovechar el ADN, que ampliará aún más los horizontes de la ciencia. Entre las posibilidades están el desarrollo de proteínas recombinantes más seguras para el tratamiento de enfermedades, la mejora de la terapia génica (43) y la producción, validación y liberación más rápida de nuevas vacunas (44). Pero, en última instancia, el potencial sólo está limitado por nuestra imaginación.
Rebecca Tirabassi es editora adjunta en Bitesizebio.com.