分子クローニングとは、組み換えDNA分子の作成を意味するようになり、生命科学全体の進歩に拍車をかけました。 1970 年代に制限エンドヌクレアーゼ (DNA の分子を選択的かつ特異的に切断する酵素) が発見されて以来、組み換え DNA 技術は応用と精巧さの両方において飛躍的に成長し、DNA 操作のためのますます強力なツールを生み出しています。 遺伝子のクローニングは非常に簡単かつ効率的に行えるようになり、今では標準的な実験手法となっています。 このため、ここ数十年の間に、遺伝子の機能に関する理解は飛躍的に深まりました。 例えば、複数のDNA断片を継ぎ目なくつなぎ合わせ、得られたプラスミドを2時間以内にバクテリアに形質転換することや、異なる構築物間で簡単に移動できるスワップ可能な遺伝子カセットを用いて、スピードと柔軟性を最大限に高めることが可能になるなど、新たな技術によってさらなる可能性が期待されている。 近い将来、分子クローニングは新しいパラダイムの出現を見ることになるだろう。合成生物学の技術により、インシリコで特定されたあらゆるDNA構築物の体外化学合成が可能になるのである。 これらの進歩により、DNA クローンの迅速な構築と反復が可能になり、遺伝子治療ベクター、組み換えタンパク質生産プロセス、新しいワクチンの開発が加速されるはずです」
Rebecca Tirabassi, Bitesize Bio.
はじめに
分子クローニングとは、任意の種から DNA 配列(多くの場合は遺伝子)を分離し、元の DNA 配列を変更せずに増殖用ベクターに挿入することを指します。 一旦単離された分子クローンは、遺伝子配列の解析のために多数のDNAコピーを作成したり、タンパク質の機能の研究または利用のために得られたタンパク質を発現させるために使用することができる。 クローンはまた、タンパク質の発現および機能を変更するためにin vitroで操作および変異させることができる。
基本的なクローニングワークフローには4つのステップが含まれる:
- 標的DNA断片(しばしば挿入物と呼ばれる)の分離
- 挿入物の適切なクローニングベクターへの結紮、組み換え分子(例えば。
- 組換えプラスミドをバクテリアまたは他の適切な宿主に形質転換して増殖させる
- 目的の組換えプラスミドを含む宿主のスクリーニング/選択
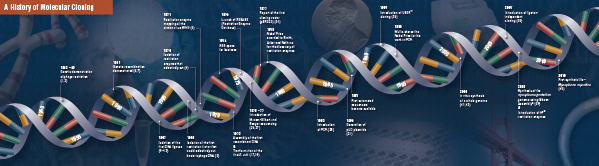
これらの画期的な4ステップは、1960年代後半から1970年代前半に複数の研究所によって慎重につなぎ合わせて行われたものである。 従来の分子クローニングを構成する発見の概要は、以下のページに記載されています。
History of Cloning
The Foundation of Molecular Cloning
Cutting (Digestion)(DIG). 組換えDNA技術は、1960年代後半に、二本鎖DNA分子を特異的に切断し結合する酵素の発見によって初めて登場した。 実は、1952年にはすでに、バクテリアが特定の宿主内でバクテリオファージの増殖を防ぐ「制限因子」をコードしていることが、2つのグループによって独立して観察されていた(1,2)。 しかし、この因子の性質が明らかになったのは、1968年、ArberとLinnが、バクテリアのDNAではなく、外来種のDNAを選択的に切断する制限因子と呼ばれる酵素の単離に成功した時であった(3)。 これらの研究はまた、細菌DNAを制限酵素から保護するメチラーゼ酵素を同定した。
ArberとLinnの発見のすぐ後、SmithはHaemophilus influenzaから制限酵素を単離し、これらの研究を拡張、確認した。 彼は、この酵素が特定の6塩基対のDNAストレッチの中央で選択的にDNAを切断することを示した。ある種の制限酵素の特徴の1つは、特定の、しばしば回文状の、「認識」配列でDNA基質を切断する性質があることである(4)。
制限酵素の威力は、制限酵素とゲル電気泳動を使ってSimian Virus 40 (SV40) ゲノムがマッピングされるまで発揮されなかった(5)。 これらの画期的な発見により、Werner Arber、Hamilton Smith、Daniel Nathansは1978年にノーベル医学賞を受賞しました。 従来のクローニングのワークフロー 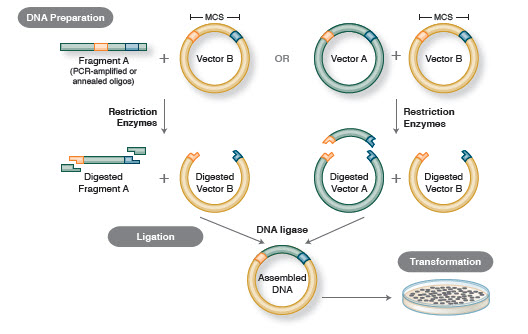
Assembling (Ligation). DNAを切断する酵素の発見と同様に、DNAを結合する酵素の発見も、それ以前の顕著な観察に先行するものであった。 1960年代初頭、2つのグループがDNA分子の切断とライゲーションによって遺伝子組み換えが起こることを発見し(6,7)、その後、バクテリオファージの直鎖DNAが宿主に感染すると急速に共有結合で閉じた円に変換されることを観察した(8)。 そのわずか2年後、5つのグループが独自にDNAリガーゼを単離し、2つのDNA断片を組み立てる能力を実証した(9-13)。
制限酵素とDNAリガーゼの発見から間もなく、最初の組換えDNA分子が作られた。 1972年、Berg はラムダファージ DNA の一部または大腸菌ガラクトースオペロンと SV40 DNA を別々に切断し、ライゲーションして最初の組み換え DNA 分子を作製した (14)。 これらの研究は、DNAが普遍的な性質を持つため、どのような生物種のDNAでも結合させることができるという概念の先駆けであった。 1980年、ポール・バーグは、「核酸の生化学、特に組み換えDNAに関する基礎的研究」に対して、ギルバート、サンガー(DNA配列決定の開発者)と共にノーベル化学賞を受賞した。
トランスフォーメーション。 新たに構築されたDNA分子を増殖させ分離する手段がなければ、組み換えDNA技術は大きく制限され、分子クローニングも不可能となる。 細菌を形質転換する能力、すなわち外来遺伝物質の取り込み、取り込み、発現を誘導する能力は、グリフィスが非致死性細菌株を熱不活性化致死性細菌と混合して致死性細菌株に転換させたときに初めて実証された(15)。 しかし、致死性をもたらす「形質転換の原理」が何であるかは、1944年までわからなかった。 同年、Avery、Macleod、McCartyは、致死性表現型を誘導するのはタンパク質ではなくDNAであることを証明した(16)。
当初、一般的な細菌実験株である大腸菌は、塩化カルシウムで大腸菌を処理するとバクテリオファージのDNAを取り込むことを証明(17)するまで形質転換に不応性だと考えられていた。 1972年、Cohenはこの原理を応用し、細菌に抗生物質耐性を与えるプラスミドを用いた形質転換の先駆者となった(18)。
基礎の構築
科学者が細菌で組換え DNA を作成および増殖する基本原則すべてを発見し適用したのに対し、このプロセスは非効率であった。 制限酵素の調製は標準化されていない精製手順のため信頼性が低く、クローニング用のプラスミドは扱いにくく、数も限られており、実験は分離できるインサート DNA の量によって制限されていた。 その後数十年にわたる研究により、分子クローニングに利用できる技術や道具が改善されました。
初期のベクター設計
最初の標準化されたベクターの開発。 ボイヤーの研究室で働く科学者たちは、一般的なクローニングプラスミド、すなわち外来 DNA のクローニングと形質転換細菌の選択のための抗生物質耐性遺伝子の発現のための固有の制限部位を持つコンパクトなプラスミドの必要性を認識しました。 1977年、彼らはクローニングを目的として設計された最初のベクター、pBR322を発表した(20)。 このベクターはサイズが4キロベースと小さく、選択用の抗生物質耐性遺伝子を2つ持っていた。
搭載されたスクリーニングとより高い収率を持つベクター。 抗生物質選択により非形質転換菌は増殖しないが、挿入DNA断片を持たずに再ライゲーションしたプラスミド(セルフライゲーション)は、細菌に抗生物質耐性を付与することが可能であった。 そのため、目的の組換えDNA分子を含む正しい細菌クローンを見つけるには時間がかかることがあった。
ヴィエイラとメッシングは、DNAが挿入されたプラスミドを含む細菌コロニーを特定するスクリーニングツールを考案した。 pBR322プラスミドをベースに、彼らは「ブルー/ホワイト・スクリーニング」システムを含む一連のpUCプラスミドを作成した(21)。 LacZ遺伝子内にいくつかのユニークな制限部位を含むマルチプルクローニングサイト(MCS)を配置することで、研究者は外来DNAを挿入したプラスミドを含む細菌コロニーをスクリーニングすることができるようになった。 pUC プラスミドは、既存のベクターよりもさらに優れており、より高いコピー数をもたらす変異を含んでいたため、プラスミドの収量が増加しました。 制限酵素を用いた初期の研究は、酵素調製の純度や各酵素に必要な緩衝液の理解不足によって妨げられた。 1975 年、New England Biolabs (NEB) 社は、組換え体から生産された制限酵素を商品化した最初の企業となった。 これにより、収率の向上、純度の改善、ロット間の一貫性、低価格化が可能になった。 現在、世界中の科学者によって、300以上の異なる配列を認識する4,000以上の制限酵素が発見されています。 NEB はまた、標準化された 4 つのバッファーシステムを開発し、このバッファーシステムにおけるすべての酵素活性を特性評価した最初の企業の 1 つです。 これにより、二重消化、つまり2つの酵素を同時に使ってDNAを消化する方法について、より深い理解を得ることができました。 その後の研究により、最も一般的な制限酵素に対応したワンバッファ・システム(NEB社のCutSmart™Bufferなど)が開発された。
配列特異性が知られている市販の制限酵素ライブラリーが登場すると、制限酵素は組み換えDNAクローンをスクリーニングするための強力なツールとなった。 診断消化」は分子クローニングで最もよく使われる技術の一つであったし、現在もそうである
ベクターと挿入物の準備。 クローニングの効率と汎用性は、ライゲーションに先立ってベクターを調製するさまざまな技術の開発によっても改善された。 アルカリ性ホスファターゼが単離され、DNA末端から3リン酸基と5リン酸基を除去することができた。 ベクターをCalf-Intestinal Phosphatase (CIP)で処理すると、DNA末端が脱リン酸化され、ベクターの自己ライゲーションを防ぎ、挿入物を持つプラスミドの回収率が高まることがすぐに発見された(24)。 リコンビナント・シュリンプ・アルカリ・ホスファターゼ(rSAP)やアンタークティック・ホスファターゼ(AP)(いずれもNEB社販売)のような熱に弱いアルカリホスファターゼの発見により、単純な温度変化でライゲーション工程の前に酵素が失活するので、工程も時間も短縮された(25)
DNAシーケンスが登場 DNAの塩基配列決定は、1970年代後半に、2つの競合する方法が考案された。 マクサムとギルバートは、DNAの化学修飾とそれに続く特定の塩基での切断に依存する「化学的配列決定法」を開発した(26)。 同時に、サンガーらは「鎖切断法」を発表し、これがほとんどの研究者に使用される方法となった(27)。 サンガー法はすぐに自動化され、1987年には最初の自動シーケンサーが販売された。
DNAのストレッチの配列を決定する能力は、分子クローニングの信頼性と多用途性を向上させた。 クローン化されると、科学者はクローンの配列を決定して、正しい組換え分子を明確に特定し、新しい遺伝子や遺伝子内の変異を特定し、追加の実験のために既知の配列に基づいてオリゴヌクレオチドを容易に設計することができました。 初期の分子クローニングにおける問題のひとつは、ベクターにクローニングするのに十分な挿入DNAを得ることであった。 1983年、Mullisはこの問題を解決する技術を考案し、分子クローニングに革命をもたらした(28)。 彼は、相反するプライマーを用いて、DNAの相補鎖を同時に増幅することにより、標的DNAの一区間を増幅した。 彼は、変性、アニーリング、重合を繰り返すことで、1本のDNAを指数関数的に増幅することを明らかにした。 ポリメラーゼ連鎖反応(PCR)は、それまで不十分だったDNAを増幅し、遺伝子をクローン化することを可能にした。 この発見により、カリー・マリスは「DNAベースの化学における手法の発展への貢献」を理由に1993年のノーベル化学賞を受賞した。
1970年にテミンとボルチモアは、RNAをDNAに変換する酵素、逆転写酵素をウイルスから独立して発見した(29、30)。 PCRが開発された直後、逆転写をPCRと結合させ(RT-PCR)、メッセンジャーRNA(mRNA)のクローニングを可能にした。 逆転写は、mRNAのDNAコピー(cDNA)を作成するために用いられ、その後、PCRによって増幅され、ライゲーション用のインサートが作成された。 この酵素の発見により、Howard Temin と David Baltimore は1975年にノーベル医学・生理学賞を受賞し、Renato Dulbecco と共有しました。 PCRの出現により、研究者はアンプリコン配列の限られた知識で遺伝子やDNA断片のクローニングができるようになった。 しかし、PCR産物をクローニングベクターに効率よくライゲーションするための最適な調製法については、ほとんどコンセンサスが得られていないのが現状であった。
当初はいくつかの異なる方法がPCR産物のクローニングに使用されていた。 PCR産物をクローニングするための最も単純な、そして現在でも最も一般的な方法は、PCR産物の末端に制限部位を導入することである(31)。 これにより、制限酵素消化後のベクターへの挿入物を直接、方向性をもってクローニングすることができる。 ブラントエンドクローニングは、ブラントエンドを生成するポリメラーゼによって生成されたPCR産物や、インサートが消化されるとブラントエンドを残すような制限部位を持つように操作されたインサートを直接ライゲーションするために開発されたものである。 これはベクターに適合する制限部位を持たないDNA断片のクローニングに有用であった(32)。
PCRの導入後まもなく、PCR産物を一つの連続したDNA配列に組み立てる方法としてオーバーラップ伸長PCRが導入された(33)。 この方法では、ベクターと重複する領域を含むPCR産物を生成するプライマーを用いて、PCRによりDNAインサートが増幅される。 その後、ベクターとインサートを混合し、変性させ、アニールさせ、インサートとベクターのハイブリダイゼーションを行う。 2回目のPCRで、インサートを含むベクターの組換えDNA分子が生成される。 オーバーラップエクステンションPCRにより、研究者は従来のPCR法では容易に増幅できなかった大きな遺伝子をつなぎ合わせることができるようになった。 オーバーラップエクステンションPCRはまた、遺伝子配列に変異を導入するために使われた(34)。
Figure 2. PCRの概要

特殊なクローニング技術の開発
分子クローニングの効率をさらに高めるために、ユニークな酵素の特性を利用したいくつかの特殊なツールや技術が開発された
TA Cloning. PCRに使用される最初の熱安定性ポリメラーゼであるTaq DNAポリメラーゼの特性を利用した方法がある。 増幅の際、Taqは各PCR産物の末端に3´dAヌクレオチドを1個付加する。 このPCR産物は、各鎖に単一のT残基を含むように切断・設計されたベクターに容易にライゲーションすることができる。 いくつかの会社がこの技術を売り出し、すでに直鎖化され「テール化」されたクローニングベクターを含むキットを販売している。
lic. ライゲーション・インディペンデント・クローニング(LIC)は、その名の通り、DNAリガーゼがない状態でDNA分子を結合させることができるものである。 LICは一般にT4 DNA Polymeraseを用いて行われ、直鎖化したベクターDNAとクローニングするインサートの両方に>12ヌクレオチド長の一本鎖DNAオーバーハングを生成させる(35)。 ベクターとインサートを混ぜ合わせると、相性の良い末端が長く伸びてアニーリングする。 この末端の長さは、リガーゼがなくても、形質転換の際にも分子を結合させるのに十分である。 一度形質転換されると、その隙間は生体内で修復される。 LIC.USERクローニングにはいくつかの異なる市販品がある。
USER クローニングは、制限酵素およびリガーゼに依存しないクローニング法として1990年代初頭に開発された(36)。 最初に考え出されたとき、この方法は、少なくとも4つのデオキシチミジン塩基がデオキシウリジンに置換された、約12ヌクレオチドの5´テールを含むPCRプライマーを使用することに依存していた。 PCR産物はウラシルDNAグリコシダーゼ(UDG)とエンドヌクレアーゼVIIIで処理され、ウラシル塩基が除去され、同様に処理したベクターにアニールできる3´オーバーラップが残るようになった。 NEBでは、リガーゼや制限酵素に依存しないクローニング反応用にUSER酵素を販売しています。
今後の動向
分子クローニングは、単一のDNA断片のクローニングから、複数のDNA成分を単一の連続したDNAに組み立てることに発展してきました。 新しい技術では、クローニングを、DNA の「ブロック」を隣り合わせに並べるような単純なプロセスに変えようとしています。 多くの新しく洗練された技術により、1本のチューブ反応内で複数のDNA断片を組み立てることができる。 これらの技術の利点は、標準化されており、シームレスで、ほとんど配列に依存しないことである。 さらに、1本のチューブで複数のDNA断片を組み立てることができるため、以前は独立していた一連の制限/ライゲーション反応が、合理的で効率的な手順へと変化する。
遺伝子アセンブリの様々な技術や製品には、SLIC (Sequence and Ligase Independent Cloning), Gibson Assembly (NEB), GeneArt® Seamless Cloning (Life Technologies) and Gateway® Cloning (Invitrogen) がある (35,37,38).
DNAアセンブリーでは、組み立てるべきDNAのブロックをPCRで増幅する。 次に、互いに隣接して組み合わされるDNA断片は、ライゲーションされる相補的な配列のブロックを含むように設計される。 これらは、ギブソンアセンブリに用いられるような互換性のある凝集末端であったり、部位特異的リコンビナーゼの認識部位を含む領域(ゲートウェイ)であったりする。 DNAライゲーションに使用される酵素は、互換性のある領域の各セットを認識し、組み立て、1回の反応で1つの連続したDNA分子を作成する。 ギブソンアッセンブリークローニング法の概要 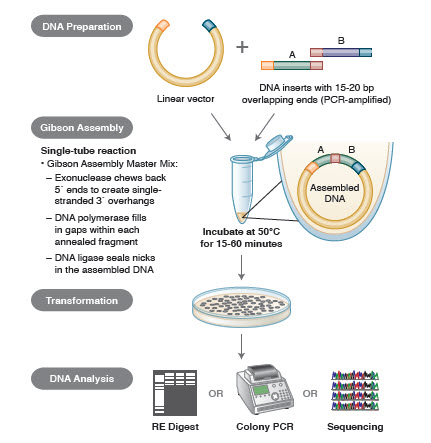
合成生物学。 DNA合成は、現在、組換えDNA技術に革命を起こしている合成生物学の分野である。 1972年に初めて完全な遺伝子が試験管内で合成されたが(40)、大きなDNA分子のDNA合成が現実のものとなったのは、研究者が全ゲノムを試験管内で合成し始めた2000年代初頭のことである(41,42)。 このような初期の実験には何年もかかったが、技術進歩により、大きなDNA分子を合成する能力は加速されている。
結論
過去40年間で、分子クローニングは、2つのDNA断片を苦労して分離してつなぎ合わせ、その後クローン候補を集中的にスクリーニングすることから、わずか数時間で最大10のDNA断片を驚くべき効率でつなぎ合わせ、あるいはDNA分子をインシリコで設計して試験管内で合成するまでに進歩した。 これらの技術により、分子生物学者はDNAを探索、操作、利用するための驚くほど強力なツールボックスを手に入れ、科学の地平をさらに広げることになる。 その可能性の中には、病気治療のためのより安全な組み換えタンパク質の開発、遺伝子治療の強化(43)、新しいワクチンの迅速な製造、検証、リリース(44)などがあります。
Rebecca Tirabassi は Bitesizebio.com の編集アシスタントです。
Molecular Cloning Technical Guide
を参照してください。