Moleculair klonen, een term die de creatie van recombinant-DNA-moleculen is gaan betekenen, heeft de vooruitgang in de hele biowetenschappen gestimuleerd. Vanaf de jaren zeventig, met de ontdekking van restrictie-endonucleasen – enzymen die selectief en specifiek DNA-moleculen doorsnijden – heeft de recombinant-DNA-technologie een exponentiële groei doorgemaakt, zowel qua toepassing als qua geavanceerdheid, met als resultaat steeds krachtigere gereedschappen voor DNA-manipulatie. Het klonen van genen is nu zo eenvoudig en efficiënt dat het een standaard laboratoriumtechniek is geworden. Dit heeft in de afgelopen decennia geleid tot een explosie van het inzicht in de genfunctie. Opkomende technologieën beloven nog meer mogelijkheden, zoals de mogelijkheid voor onderzoekers om meerdere DNA-fragmenten naadloos aan elkaar te hechten en de resulterende plasmiden in bacteriën te transformeren, in minder dan twee uur, of het gebruik van verwisselbare gencassettes, die gemakkelijk tussen verschillende constructies kunnen worden verplaatst, om de snelheid en flexibiliteit te maximaliseren. In de nabije toekomst zal voor moleculair klonen waarschijnlijk een nieuw paradigma opduiken, met technieken uit de synthetische biologie die in vitro chemische synthese van elk in silico gespecificeerd DNA-construct mogelijk zullen maken. Deze vooruitgang zou een snellere constructie en iteratie van DNA-klonen mogelijk moeten maken, waardoor de ontwikkeling van gentherapievectoren, recombinante eiwitproductieprocessen en nieuwe vaccins wordt versneld.
Rebecca Tirabassi, Bitesize Bio.
Inleiding
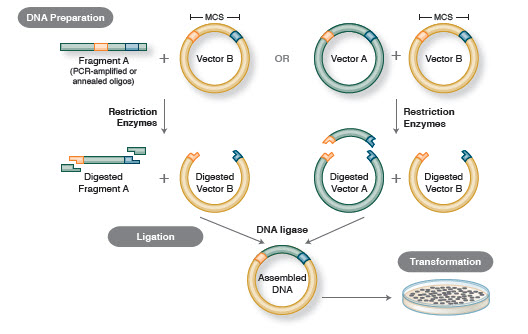
Moleculair klonen verwijst naar de isolatie van een DNA-sequentie van een soort (vaak een gen), en de invoeging daarvan in een vector voor vermeerdering, zonder wijziging van de oorspronkelijke DNA-sequentie. Eenmaal geïsoleerd kunnen moleculaire klonen worden gebruikt om vele kopieën van het DNA te genereren voor analyse van de gensequentie, en/of om het resulterende eiwit tot expressie te brengen voor het bestuderen of benutten van de functie van het eiwit. De klonen kunnen ook in vitro worden gemanipuleerd en gemuteerd om de expressie en functie van het eiwit te veranderen.
De basisworkflow voor klonen omvat vier stappen:
- Isolatie van DNA-doelfragmenten (vaak inserts genoemd)
- Ligatie van inserts in een geschikte kloneringsvector, waardoor recombinante moleculen worden gecreëerd (bv, plasmiden)
- Transformatie van recombinante plasmiden in bacteriën of andere geschikte gastheren voor vermeerdering
- Screening/selectie van gastheren die het beoogde recombinante plasmide bevatten
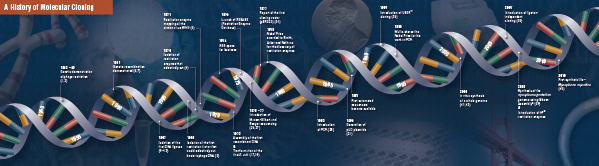
Deze vier baanbrekende stappen werden zorgvuldig samengevoegd en uitgevoerd door meerdere laboratoria, beginnend aan het eind van de jaren zestig en het begin van de jaren zeventig. Een samenvatting van de ontdekkingen die het traditionele moleculaire klonen omvatten, wordt op de volgende pagina’s beschreven.
Geschiedenis van het klonen
De basis van het moleculaire klonen
Snijden (Digestie). De recombinant-DNA-technologie kwam aan het eind van de jaren zestig op, met de ontdekking van enzymen die specifiek dubbelstrengs DNA-moleculen konden knippen en verbinden. Reeds in 1952 werd door twee groepen onafhankelijk van elkaar waargenomen dat bacteriën codeerden voor een “beperkingsfactor” die verhinderde dat bacteriofagen in bepaalde gastheren konden groeien (1,2). De aard van deze factor werd echter pas in 1968 ontdekt, toen Arber en Linn erin slaagden een enzym te isoleren, restrictiefactor genoemd, dat selectief exogeen DNA afsneed, maar niet bacterieel DNA (3). In deze studies werd ook een methylase-enzym geïdentificeerd dat het bacteriële DNA tegen restrictie-enzymen beschermde.
Kort na de ontdekking van Arber en Linn breidde Smith deze studies uit en bevestigde ze door een restrictie-enzym van Haemophilus influenza te isoleren. Hij toonde aan dat het enzym selectief DNA sneed in het midden van een specifiek stuk DNA van 6 basenparen; een kenmerk van bepaalde restrictie-enzymen is hun neiging om het DNA-substraat in of nabij specifieke, vaak palindromische, “herkennings”-sequenties te snijden (4).
De volledige kracht van restrictie-enzymen werd pas gerealiseerd toen restrictie-enzymen en gel-elektroforese werden gebruikt om het genoom van het Simian Virus 40 (SV40) in kaart te brengen (5). Voor deze baanbrekende bevindingen deelden Werner Arber, Hamilton Smith en Daniel Nathans in 1978 de Nobelprijs voor geneeskunde.
Figuur 1. Traditionele kloneringsworkflow
Assembling (Ligation). Net als de ontdekking van enzymen die DNA doorsnijden, werd de ontdekking van een enzym dat DNA kan samenvoegen voorafgegaan door eerdere, saillante observaties. In het begin van de jaren zestig ontdekten twee groepen dat genetische recombinatie kon plaatsvinden door het breken en binden van DNA-moleculen (6,7), op de voet gevolgd door de waarneming dat lineair bacteriofaag-DNA snel wordt omgezet in covalent gesloten cirkels na infectie van de gastheer (8). Slechts twee jaar later isoleerden vijf groepen onafhankelijk van elkaar DNA-ligases en toonden aan dat zij in staat zijn twee stukken DNA te assembleren (9-13).
Nauwelijks na de ontdekking van restrictie-enzymen en DNA-ligasen werd de eerste recombinant-DNA-molecule gemaakt. In 1972 sneed Berg afzonderlijk een stuk lambda bacteriofaag-DNA of het E. coli galactose operon door en ligeerde dit met SV40-DNA om de eerste recombinant-DNA-moleculen te maken (14). Deze studies waren de wegbereiders van het concept dat, door de universele aard van DNA, DNA van om het even welke soort kan worden samengevoegd. In 1980 deelde Paul Berg de Nobelprijs voor Scheikunde met Walter Gilbert en Frederick Sanger (de ontwikkelaars van DNA-sequencing), voor “zijn fundamentele studies van de biochemie van nucleïnezuren, met bijzondere aandacht voor recombinant-DNA”.
Transformatie. De recombinant-DNA-technologie zou ernstig worden beperkt, en moleculair klonen onmogelijk, zonder de middelen om de nieuw geconstrueerde DNA-molecule te vermeerderen en te isoleren. Het vermogen om bacteriën te transformeren, of de opname, opname en expressie van vreemd genetisch materiaal te induceren, werd voor het eerst aangetoond door Griffith toen hij een niet-dodelijke bacteriestam transformeerde in een dodelijke stam door de niet-dodelijke stam te mengen met door warmte geïnactiveerde dodelijke bacteriën (15). De aard van het “omvormingsprincipe” dat de dodelijkheid overbrengt, werd echter pas in 1944 begrepen. In datzelfde jaar toonden Avery, Macleod en McCarty aan dat DNA, en niet eiwit, verantwoordelijk was voor het induceren van het dodelijke fenotype (16).
In eerste instantie dacht men dat de gewone bacteriële laboratoriumstam, E. coli, ongevoelig was voor transformatie, totdat Mandel en Higa aantoonden dat behandeling van E. coli met calciumchloride de opname van bacteriofaag-DNA induceerde (17). Cohen paste dit principe toe toen hij in 1972 een pioniersrol vervulde bij de transformatie van bacteriën met plasmiden om de bacteriën antibioticaresistentie te verlenen (18).
Het ultieme experiment: digestie, ligatie en transformatie van een recombinant DNA-molecuul werd uitgevoerd door Boyer, Cohen en Chang in 1973, toen zij het plasmide pSC101 met EcoRI verteerden, het gelineariseerde fragment aan een ander enzym-restricted plasmide ligeerden en het resulterende recombinant molecuul in E. coli, waardoor de bacteriën resistent werden tegen tetracycline (19), waarmee de basis werd gelegd voor het meeste recombinant-DNA-werk sindsdien.
Bouwen op de basis
Weliswaar hadden wetenschappers alle basisprincipes voor het maken en vermeerderen van recombinant-DNA in bacteriën ontdekt en toegepast, maar het proces was inefficiënt. Bereidingen van restrictie-enzymen waren onbetrouwbaar door niet-gestandaardiseerde zuiveringsprocedures, plasmiden voor klonering waren omslachtig, moeilijk om mee te werken en beperkt in aantal, en experimenten werden beperkt door de hoeveelheid insert-DNA die kon worden geïsoleerd. Onderzoek gedurende de volgende decennia leidde tot verbeteringen in de technieken en hulpmiddelen die beschikbaar waren voor moleculair klonen.
Eerste vectorontwerp.
Ontwikkeling van de eerste gestandaardiseerde vector. Wetenschappers die in het laboratorium van Boyer werkten, onderkenden de behoefte aan een algemeen kloneringsplasmide, een compact plasmide met unieke restrictieplaatsen voor het kloneren in vreemd DNA en de expressie van antibiotica-resistentiegenen voor de selectie van getransformeerde bacteriën. In 1977 beschreven zij de eerste vector die voor kloneringsdoeleinden was ontworpen, pBR322 (20). Deze vector was klein, ~4 kilobasen groot, en had twee antibiotica-resistentiegenen voor selectie.
Vectoren met on-board screening en hogere opbrengsten. Hoewel antibioticaselectie verhinderde dat niet-getransformeerde bacteriën groeiden, konden plasmiden die re-ligeerden zonder insert-DNA-fragmenten (zelf-ligatie) toch antibioticaresistentie aan bacteriën verlenen. Daarom kon het vinden van de juiste bacteriële klonen die het gewenste recombinant-DNA-molecuul bevatten, tijdrovend zijn.
Vieira en Messing bedachten een screeninginstrument om bacteriële kolonies te identificeren die plasmiden met DNA-inserts bevatten. Op basis van het pBR322 plasmide creëerden zij de reeks pUC plasmiden, die een “blauw/wit screening” systeem bevatte (21). Door de plaatsing van een meervoudige kloneringsplaats (MCS) die verschillende unieke restrictieplaatsen binnen het LacZ´-gen bevat, konden de onderzoekers bacteriekolonies screenen die plasmiden met het vreemde DNA-insert bevatten. Wanneer bacteriën op de juiste media werden uitgezet, bevatten de witte kolonies plasmiden met inserts, terwijl de blauwe kolonies plasmiden zonder inserts bevatten. pUC plasmiden hadden een bijkomend voordeel ten opzichte van bestaande vectoren; zij bevatten een mutatie die resulteerde in hogere kopie-aantallen, waardoor de plasmide-opbrengst toenam.
Verbetering van restrictie-ontsluitingen. Het vroege werk met restrictie-enzymen werd gehinderd door de zuiverheid van het enzympreparaat en een gebrek aan inzicht in de buffervereisten voor elk enzym. In 1975 was New England Biolabs (NEB) het eerste bedrijf dat restrictie-enzymen op de markt bracht die van recombinante oorsprong waren. Dit maakte een hogere opbrengst, een betere zuiverheid, consistentie van partij tot partij en een lagere prijs mogelijk. Momenteel zijn er meer dan 4.000 restrictie-enzymen, die meer dan 300 verschillende sequenties herkennen, ontdekt door wetenschappers over de hele wereld. NEB levert momenteel meer dan 230 van deze specificiteiten.
NEB was ook een van de eerste bedrijven die een gestandaardiseerd vier-buffersysteem ontwikkelde, en om al zijn enzymactiviteiten in dit buffersysteem te karakteriseren. Dit leidde tot een beter begrip van het uitvoeren van een dubbele digestie, of de digestie van DNA met twee enzymen sim-gelijktijdig. Later onderzoek leidde tot de ontwikkeling van één-buffersystemen, die compatibel zijn met de meest voorkomende restrictie-enzymen (zoals NEB’s CutSmart™ Buffer).
Met de komst van commercieel verkrijgbare restrictie-enzymenbibliotheken met bekende sequentiespecificiteiten werden restrictie-enzymen een krachtig hulpmiddel voor het screenen van potentiële recombinant-DNA-klonen. De “diagnostische digestie” was, en is nog steeds, een van de meest gebruikte technieken bij het moleculair klonen.
Vector- en insertvoorbereiding. De kloneringsefficiëntie en -veelzijdigheid werden ook verbeterd door de ontwikkeling van verschillende technieken voor de voorbereiding van vectoren vóór de ligering. Alkalische fosfatasen werden geïsoleerd die de 3´ en 5´ fosfaatgroepen van de uiteinden van het DNA konden verwijderen. Al snel werd ontdekt dat behandeling van vectoren met kalfsintestinaal fosfatase (CIP) de DNA-einden defosforyleerde en zelf-ligatie van de vector verhinderde, waardoor meer plasmiden met insert werden teruggevonden (24).
Het CIP-enzym bleek moeilijk te inactiveren, en elke resterende activiteit leidde tot defosforylering van insert-DNA en remming van de ligatiereactie. De ontdekking van de hitte-labiele alkalische fosfatasen, zoals recombinant Garnalen Alkalische Fosfatase (rSAP) en Antarctische Fosfatase (AP) (beide verkocht door NEB), verminderde de betrokken stappen en tijd, aangezien een eenvoudige verschuiving in temperatuur het enzym voorafgaand aan de ligatiestap inactiveert (25).
DNA-sequencing arriveert. DNA-sequencing werd ontwikkeld aan het eind van de jaren 1970, toen twee concurrerende methoden werden bedacht. Maxam en Gilbert ontwikkelden de “chemische sequencingmethode”, die berustte op chemische modificatie van DNA en daaropvolgende splitsing op specifieke basen (26). Tegelijkertijd publiceerden Sanger en collega’s over de “ketting-terminatiemethode”, die de door de meeste onderzoekers gebruikte methode werd (27). De Sanger-methode werd al snel geautomatiseerd, en de eerste automatische sequencers werden in 1987 verkocht.
De mogelijkheid om de sequentie van een stuk DNA te bepalen verbeterde de betrouwbaarheid en veelzijdigheid van moleculair klonen. Eenmaal gekloond, konden wetenschappers klonen sequencen om definitief de juiste recombinante molecule te identificeren, nieuwe genen of mutaties in genen te identificeren, en gemakkelijk oligonucleotiden ontwerpen op basis van de bekende sequentie voor aanvullende experimenten.
De impact van de polymerasekettingreactie. Een van de problemen bij het moleculaire klonen in de beginjaren was het verkrijgen van voldoende insert-DNA om in de vector te klonen. In 1983 bedacht Mullis een techniek die dit probleem oploste en een revolutie teweegbracht in het moleculaire klonen (28). Hij amplificeerde een stuk doel-DNA door tegengestelde primers te gebruiken om beide complementaire DNA-strengen tegelijk te amplificeren. Door cycli van denaturatie, annealing en polymerisatie toonde hij aan dat hij een enkele kopie van DNA exponentieel kon amplificeren. De polymerase kettingreactie, of PCR, maakte het mogelijk genen te amplificeren en te klonen uit voorheen ontoereikende hoeveelheden DNA. Voor deze ontdekking kreeg Kary Mullis in 1993 de Nobelprijs voor de scheikunde “voor zijn bijdragen aan de ontwikkeling van methoden binnen de chemie op basis van DNA”.
In 1970 ontdekten Temin en Baltimore onafhankelijk van elkaar reverse transcriptase in virussen, een enzym dat RNA omzet in DNA (29,30). Kort nadat PCR was ontwikkeld, werd omgekeerde transcriptie gekoppeld aan PCR (RT-PCR) om het klonen van boodschapper-RNA (mRNA) mogelijk te maken. Omgekeerde transcriptie werd gebruikt om een DNA-kopie (cDNA) van mRNA te maken dat vervolgens door PCR werd geamplificeerd om een insert te maken voor ligatie. Voor hun ontdekking van het enzym kregen Howard Temin en David Baltimore in 1975 de Nobelprijs voor geneeskunde en fysiologie, die zij deelden met Renato Dulbecco.
Clonering van PCR-producten. De komst van PCR betekende dat onderzoekers nu genen en DNA-segmenten konden klonen met beperkte kennis van de sequentie van het amplicon. Er bestond echter weinig eensgezindheid over de optimale methode voor de bereiding van PCR-producten met het oog op een efficiënte ligering in kloneringsvectoren.
Voor het klonen van PCR-producten werden aanvankelijk verschillende methoden gebruikt. De eenvoudigste en nog steeds de meest gebruikte methode voor het klonen van PCR-producten is de invoering van restrictieplaatsen aan de uiteinden van het PCR-product (31). Dit maakt een directe, gerichte klonering van het insert in de vector na restrictie digestie mogelijk. Blunt-ended klonering werd ontwikkeld om PCR-producten gegenereerd door polymerasen die stompe uiteinden geproduceerd, of inserts gemanipuleerd om restrictieplaatsen die stompe uiteinden links zodra het inzetstuk werd verteerd hebben direct ligeren. Dit was nuttig voor het klonen van DNA-fragmenten die geen restrictieplaatsen bevatten die compatibel zijn met de vector (32).
Kort na de introductie van PCR werd overlap-extensie-PCR geïntroduceerd als een methode om PCR-producten tot één aaneengesloten DNA-sequentie samen te voegen (33). Bij deze methode wordt het DNA-insert geamplificeerd door PCR met gebruikmaking van primers die een PCR-product genereren dat overlappende regio’s met de vector bevat. De vector en de insert worden vervolgens gemengd, gedenatureerd en geannealed, waardoor hybridisatie van de insert aan de vector mogelijk wordt. Een tweede ronde van PCR genereert recombinant-DNA-moleculen van de insert-bevattende vector. Overlap extension PCR stelde onderzoekers in staat grote genen samen te stellen die niet gemakkelijk met traditionele PCR-methoden konden worden geamplificeerd. Overlap extension PCR werd ook gebruikt om mutaties in gensequenties aan te brengen (34).
Figuur 2. Overzicht van PCR

Ontwikkeling van gespecialiseerde kloneringstechnieken.
In een poging om de efficiëntie van moleculair kloneren verder te verbeteren, werden verschillende gespecialiseerde hulpmiddelen en technieken ontwikkeld die gebruik maakten van de eigenschappen van unieke enzymen.
TA-klonering. Bij één benadering werd gebruik gemaakt van een eigenschap van Taq DNA-polymerase, het eerste hittestabiele polymerase dat voor PCR werd gebruikt. Tijdens de amplificatie voegt Taq een enkele 3´ dA nucleotide toe aan het eind van elk PCR-product. Het PCR-product kan gemakkelijk worden geligeerd in een vector die zo is gesneden en gemodificeerd dat hij op elke streng enkele T-residuen bevat. Verscheidene bedrijven hebben deze techniek op de markt gebracht en verkopen kits met kloneringsvectoren die reeds zijn gelineariseerd en “tailed”.
LIC. Ligatie-onafhankelijk klonen (LIC) maakt, zoals de naam al aangeeft, de verbinding van DNA-moleculen mogelijk in afwezigheid van DNA-ligase. LIC wordt gewoonlijk uitgevoerd met T4 DNA-polymerase, dat wordt gebruikt om enkelstrengs DNA-overhangs te genereren, >12 nucleotiden lang, op zowel het gelineariseerde vector-DNA als het te kloneren insert (35). Wanneer de vector en het insert worden samengevoegd, annealiseren zij door het lange stuk van compatibele uiteinden. De lengte van de compatibele uiteinden is voldoende om de molecule bij elkaar te houden in afwezigheid van ligase, zelfs tijdens de transformatie. Eenmaal getransformeerd, worden de gaten in vivo gerepareerd. Er zijn verschillende commercieel verkrijgbare producten voor LIC.USER klonering.
USER klonering werd voor het eerst ontwikkeld in de vroege jaren 1990 als een restrictie-enzym- en ligase-onafhankelijke kloneringsmethode (36). Toen de methode voor het eerst werd ontwikkeld, was deze gebaseerd op het gebruik van PCR-primers die een 5´-staart van ~12 nucleotiden bevatten, waarin ten minste vier deoxythymidinebasen waren vervangen door deoxyuridines. Het PCR-product werd behandeld met uracil-DNA-glycosidase (UDG) en endonuclease VIII, dat de uracilbasen wegsnijdt en een 3´-overlap overlaat die aan een op dezelfde wijze behandelde vector kan worden ge¨nealed. NEB verkoopt het USER-enzym voor ligase- en restrictie-enzymonafhankelijke kloneringsreacties.
Toekomsttrends
Moleculair kloneren heeft zich ontwikkeld van het kloneren van een enkel DNA-fragment tot het assembleren van meerdere DNA-componenten in een enkel aaneengesloten stuk DNA. Nieuwe en opkomende technologieën trachten het klonen om te vormen tot een proces dat zo eenvoudig is als het naast elkaar rangschikken van “blokken” DNA.
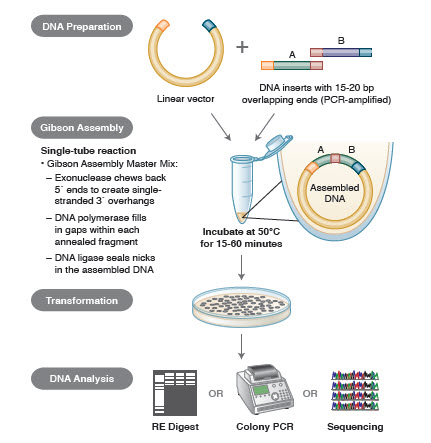
DNA-assemblagemethoden. Veel nieuwe, elegante technologieën maken de assemblage van meerdere DNA-fragmenten in een enkele buisreactie mogelijk. De voordelen van deze technologieën zijn dat zij gestandaardiseerd, naadloos en grotendeels sequentie-onafhankelijk zijn. Bovendien verandert de mogelijkheid om meerdere DNA-fragmenten in één buis te assembleren een reeks voorheen onafhankelijke restrictie-/rigatiereacties in een gestroomlijnde, efficiënte procedure.
Verschillende technieken en producten voor genassemblage omvatten SLIC (Sequence and Ligase Independent Cloning), Gibson Assembly (NEB), GeneArt® Seamless Cloning (Life Technologies) en Gateway® Cloning (Invitrogen) (35,37,38).
Bij DNA-assemblage worden de te assembleren DNA-blokken met behulp van PCR geamplificeerd. Vervolgens worden de DNA-fragmenten die naast elkaar moeten worden geassembleerd, zodanig gemanipuleerd dat zij blokken complementaire sequenties bevatten die aan elkaar zullen worden geligeerd. Dit kunnen compatibele cohesieve uiteinden zijn, zoals die welke voor Gibson-assemblage worden gebruikt, of gebieden die herkenningspunten bevatten voor plaatsspecifieke recombinasen (Gateway). Het enzym dat voor DNA-ligatie wordt gebruikt, herkent en assembleert elke set van compatibele gebieden, waardoor in één reactie één aaneengesloten DNA-molecuul ontstaat.
Figuur 3. Overzicht van de Gibson Assembly-kloneringsmethode
Synthetische biologie. DNA-synthese is een gebied van de synthetische biologie dat momenteel een revolutie teweegbrengt in de recombinant-DNA-technologie. Hoewel in 1972 voor het eerst een volledig gen in vitro werd gesynthetiseerd (40), werd DNA-synthese van grote DNA-moleculen pas een realiteit in het begin van de jaren 2000, toen onderzoekers hele genomen in vitro begonnen te synthetiseren (41,42). Deze vroege experimenten duurde jaren om te voltooien, maar de technologie versnelt de mogelijkheid om grote DNA-moleculen te synthetiseren.
Conclusie
In de afgelopen 40 jaar heeft moleculair klonen zich ontwikkeld van het moeizaam isoleren en samenvoegen van twee stukken DNA, gevolgd door intensieve screening van potentiële klonen, tot het naadloos samenvoegen van maximaal 10 DNA-fragmenten met opmerkelijke efficiëntie in slechts een paar uur, of het ontwerpen van DNA-moleculen in silico en het synthetiseren ervan in vitro. Samen bieden al deze technologieën moleculaire biologen een verbazingwekkend krachtig instrumentarium voor het onderzoeken, manipuleren en benutten van DNA, dat de horizon van de wetenschap verder zal verruimen. Tot de mogelijkheden behoren de ontwikkeling van veiliger recombinante proteïnen voor de behandeling van ziekten, de verbetering van gentherapie (43) en een snellere productie, validatie en vrijgave van nieuwe vaccins (44). Maar uiteindelijk wordt het potentieel alleen beperkt door onze verbeelding.
Rebecca Tirabassi is een assistent-redacteur bij Bitesizebio.com.