Clonagem molecular, um termo que chegou a significar a criação de moléculas de DNA recombinantes, tem estimulado o progresso através das ciências da vida. A partir dos anos 70, com a descoberta das endonucleases restritivas – enzimas que seletiva e especificamente cortam moléculas de DNA – a tecnologia do DNA recombinante tem visto um crescimento exponencial tanto na aplicação quanto na sofisticação, produzindo ferramentas cada vez mais poderosas para a manipulação do DNA. A clonagem de genes é agora tão simples e eficiente que se tornou uma técnica laboratorial padrão. Isto levou a uma explosão na compreensão da função dos genes nas últimas décadas. Tecnologias emergentes prometem possibilidades ainda maiores, tais como permitir aos pesquisadores costurar sem problemas múltiplos fragmentos de DNA e transformar os plasmídeos resultantes em bactérias, em menos de duas horas, ou o uso de cassetes de genes intercambiáveis, que podem ser facilmente movidos entre diferentes construções, para maximizar a velocidade e a flexibilidade. Num futuro próximo, a clonagem molecular irá provavelmente assistir ao surgimento de um novo paradigma, com técnicas de biologia sintética que irão permitir a síntese química in vitro de qualquer construção de ADN em silício especificado. Estes avanços deverão permitir uma construção e iteração mais rápida dos clones de DNA, acelerando o desenvolvimento de vetores de terapia genética, processos de produção de proteínas recombinantes e novas vacinas.
Rebecca Tirabassi, Bitesize Bio.
Introdução
Clonagem molecular refere-se ao isolamento de uma seqüência de DNA de qualquer espécie (muitas vezes um gene), e sua inserção em um vetor de propagação, sem alteração da seqüência original do DNA. Uma vez isolados, os clones moleculares podem ser usados para gerar muitas cópias do DNA para análise da sequência genética, e/ou para expressar a proteína resultante para o estudo ou utilização da função da proteína. Os clones também podem ser manipulados e mutados in vitro para alterar a expressão e função da proteína.
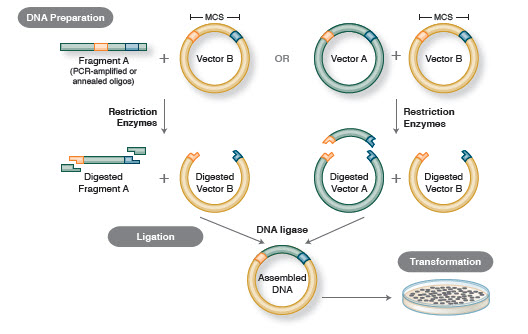
O fluxo de trabalho básico de clonagem inclui quatro etapas:
- Isolamento de fragmentos de DNA alvo (frequentemente referidos como inserções)
- Ligação de inserções em um vetor de clonagem apropriado, criando moléculas recombinantes (por exemplo plasmídeos)
- Transformação de plasmídeos recombinantes em bactérias ou outro hospedeiro adequado para propagação
- Triagem/selecção de hospedeiros contendo o plasmídeo recombinante pretendido
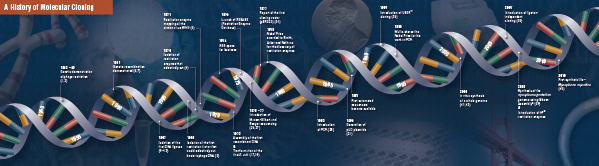
Estas quatro etapas revolucionárias foram cuidadosamente unidas e realizadas por vários laboratórios, começando no final dos anos 60 e início dos anos 70. Um resumo das descobertas que compõem a clonagem molecular tradicional é descrito nas páginas seguintes.
História da Clonagem
The Foundation of Molecular Cloning
Corte (Digestão). A tecnologia do DNA Recombinante surgiu no final dos anos 60, com a descoberta de enzimas que podiam especificamente cortar e unir moléculas de DNA de dupla cadeia. De facto, já em 1952, dois grupos observaram independentemente que as bactérias codificavam um “factor de restrição” que impedia o crescimento de bacteriófagos dentro de certos hospedeiros (1,2). Mas a natureza do fator não foi descoberta até 1968, quando Arber e Linn conseguiram isolar uma enzima, chamada fator de restrição, que cortou seletivamente o DNA exógeno, mas não o DNA bacteriano (3). Estes estudos também identificaram uma enzima de metilase que protegia o DNA bacteriano das enzimas de restrição.
Pouco depois da descoberta de Arber e Linn, Smith ampliou e confirmou estes estudos isolando uma enzima de restrição do Haemophilus influenza. Ele demonstrou que a enzima cortou seletivamente o DNA no meio de uma extensão específica de 6 pares de base de DNA; uma característica de certas enzimas de restrição é sua propensão para cortar o substrato de DNA em ou perto de seqüências específicas, freqüentemente palíndromas, de “reconhecimento” (4).
O poder total das enzimas de restrição não foi realizado até que as enzimas de restrição e eletroforese em gel fossem usadas para mapear o genoma do Vírus Simiano 40 (SV40) (5). Para estes achados seminais, Werner Arber, Hamilton Smith e Daniel Nathans compartilharam o Prêmio Nobel de Medicina de 1978.
Figure 1. Traditional Cloning Workflow
Assembling (Ligation). Assim como a descoberta de enzimas que cortam o DNA, a descoberta de uma enzima que poderia unir o DNA foi precedida por observações anteriores e salientes. No início dos anos 60, dois grupos descobriram que a recombinação genética poderia ocorrer através da quebra e ligação de moléculas de DNA (6,7), seguida de perto pela observação de que o DNA bacteriófago linear é rapidamente convertido em círculos covalentemente fechados após a infecção do hospedeiro (8). Apenas dois anos depois, cinco grupos isolaram independentemente as ligaduras de ADN e demonstraram a sua capacidade de reunir dois pedaços de ADN (9-13).
Não muito tempo após a descoberta das enzimas de restrição e ligas de DNA, a primeira molécula de DNA recombinante foi feita. Em 1972, Berg cortou e ligou separadamente um pedaço de DNA bacteriófago lambda ou o operon E. coli galactose com DNA SV40 para criar as primeiras moléculas recombinantes de DNA (14). Estes estudos foram pioneiros no conceito de que, devido à natureza universal do DNA, o DNA de qualquer espécie poderia ser unido. Em 1980, Paul Berg compartilhou o Prêmio Nobel de Química com Walter Gilbert e Frederick Sanger (os desenvolvedores do sequenciamento de DNA), por “seus estudos fundamentais da bioquímica dos ácidos nucléicos, com particular atenção ao DNA recombinante”.
Transformação. A tecnologia do DNA recombinante seria severamente limitada, e a clonagem molecular impossível, sem os meios para propagar e isolar a molécula de DNA recém-construída. A capacidade de transformar bactérias, ou induzir a absorção, incorporação e expressão de material genético estranho, foi demonstrada pela primeira vez por Griffith quando ele transformou uma estirpe não letal de bactérias em uma estirpe letal, misturando a estirpe não letal com bactérias letais ativadas por calor (15). Contudo, a natureza do “princípio transformador” que transmitia a letalidade não foi entendida até 1944. No mesmo ano, Avery, Macleod e McCarty demonstraram que o DNA, e não a proteína, era responsável pela indução do fenótipo letal (16).
Inicialmente, acreditava-se que a estirpe comum de laboratório bacteriano, E. coli, era refratária à transformação, até que Mandel e Higa demonstraram que o tratamento da E. coli com cloreto de cálcio induziu a absorção do DNA bacteriófago (17). Cohen aplicou este princípio, em 1972, quando foi pioneiro na transformação de bactérias com plasmídeos para conferir resistência antibiótica às bactérias (18).
O experimento final: digestão, ligação e transformação de uma molécula de DNA recombinante foi executado por Boyer, Cohen e Chang em 1973, quando digeriram o plasmídeo pSC101 com EcoRI, ligaram o fragmento linearizado a outro plasmídeo com restrição enzimática e transformaram a molécula recombinante resultante em E. coli, conferindo resistência tetraciclina à bactéria (19), estabelecendo assim a base para a maioria dos trabalhos de DNA recombinante desde.
Construindo a base
Embora os cientistas tivessem descoberto e aplicado todos os princípios básicos para criar e propagar DNA recombinante em bactérias, o processo era ineficiente. As preparações enzimáticas de restrição não eram confiáveis devido a procedimentos de purificação não padronizados, os plasmídeos para clonagem eram pesados, difíceis de trabalhar e em número limitado, e as experiências eram limitadas pela quantidade de DNA inserido que podia ser isolado. As pesquisas das décadas seguintes levaram a melhorias nas técnicas e ferramentas disponíveis para clonagem molecular.
Desenho do primeiro vetor padronizado.
Desenvolvimento do primeiro vetor padronizado. Os cientistas que trabalham no laboratório da Boyer reconheceram a necessidade de um plasmídeo de clonagem geral, um plasmídeo compacto com locais de restrição únicos para clonagem em DNA estranho e a expressão de genes de resistência a antibióticos para seleção de bactérias transformadas. Em 1977, eles descreveram o primeiro vetor projetado para fins de clonagem, pBR322 (20). Este vetor era pequeno, ~4 kilobases de tamanho, e tinha dois genes de resistência a antibióticos para seleção.
Vetores com triagem a bordo e maiores rendimentos. Embora a seleção antibiótica tenha impedido o crescimento de bactérias não transformadas, plasmídeos que se re-aligavam sem fragmentos de DNA inserido (auto-ligação) ainda poderiam conferir resistência antibiótica às bactérias. Portanto, encontrar os clones bacterianos corretos contendo a molécula de DNA recombinante desejada poderia ser demorado.
Vieira e Messing criaram uma ferramenta de triagem para identificar colônias bacterianas contendo plasmídeos com inserções de DNA. Com base no plasmídeo pBR322, eles criaram a série de plasmídeos pUC, que continham um sistema de “triagem azul/branco” (21). A colocação de um local de clonagem múltipla (MCS) contendo vários locais de restrição únicos dentro do gene LacZ’ permitiu aos pesquisadores fazer a triagem de colônias bacterianas contendo plasmídeos com a inserção de DNA estranho. Quando as bactérias eram plaqueadas na mídia correta, as colônias brancas continham plasmídeos com inserções, enquanto as colônias azuis continham plasmídeos sem inserções. Os plasmídeos pUC tinham uma vantagem adicional sobre os vetores existentes; eles continham uma mutação que resultava em números de cópias mais altos, portanto aumentando o rendimento de plasmídeos.
Melhorando os digestores de restrição. O trabalho precoce com enzimas de restrição foi prejudicado pela pureza do preparado enzimático e pela falta de compreensão dos requisitos de tampão para cada enzima. Em 1975, a New England Biolabs (NEB) tornou-se a primeira empresa a comercializar enzimas de restrição produzidas a partir de uma fonte recombinante. Isto permitiu maiores rendimentos, melhor pureza, consistência lote a lote e preços mais baixos. Atualmente, mais de 4.000 enzimas de restrição, reconhecendo mais de 300 sequências diferentes, foram descobertas por cientistas em todo o mundo. NEB atualmente fornece mais de 230 destas especificidades.
NEB foi também uma das primeiras empresas a desenvolver um sistema de quatro tampão padronizado, e a caracterizar todas as suas atividades enzimáticas neste sistema tampão. Isto levou a um melhor entendimento de como conduzir uma digestão dupla, ou a digestão do DNA com duas enzimas simultaneamente. Pesquisas posteriores levaram ao desenvolvimento de sistemas de um tampão, que são compatíveis com as enzimas de restrição mais comuns (como o tampão CutSmart™ da NEB).
Com o advento das bibliotecas de enzimas de restrição disponíveis comercialmente com especificidades de sequência conhecidas, as enzimas de restrição tornaram-se uma ferramenta poderosa para a triagem de potenciais clones de DNA recombinantes. A “digestão diagnóstica” foi, e ainda é, uma das técnicas mais comuns usadas na clonagem molecular.
Vetor e preparação de insertos. A eficiência e versatilidade da clonagem também foram melhoradas pelo desenvolvimento de diferentes técnicas de preparação de vetores antes da ligadura. Foram isoladas fosfatases alcalinas que podiam remover os grupos fosfato de 3′ e 5′ das extremidades do DNA . Logo foi descoberto que o tratamento dos vetores com fosfatase califosfatase intestinal (CIP) desfosforada de DNA e evitou a auto-ligação do vetor, aumentando a recuperação dos plasmídeos com insert (24).
A enzima CIP mostrou-se difícil de inativar, e qualquer atividade residual levou à desfosforilação do DNA insert e inibição da reação de ligadura. A descoberta das fosfatases alcalinas termo-lábeis, como a fosfatase alcalina recombinante do camarão (rSAP) e a fosfatase antártica (AP) (ambas vendidas pela NEB), diminuiu as etapas e o tempo envolvido, pois uma simples mudança de temperatura inativa a enzima antes da etapa de ligadura (25).
Chega a sequenciação do ADN. O sequenciamento de DNA foi desenvolvido no final da década de 1970, quando dois métodos concorrentes foram concebidos. Maxam e Gilbert desenvolveram o “método de sequenciamento químico”, que se baseou na modificação química do DNA e subsequente clivagem em bases específicas (26). Ao mesmo tempo, Sanger e colegas publicaram sobre o “método de terminação em cadeia”, que se tornou o método utilizado pela maioria dos pesquisadores (27). O método Sanger tornou-se rapidamente automatizado, e os primeiros sequenciadores automáticos foram vendidos em 1987.
A capacidade de determinar a sequência de um trecho de DNA aumentou a confiabilidade e versatilidade da clonagem molecular. Uma vez clonados, os cientistas puderam sequenciar clones para identificar definitivamente a molécula recombinante correta, identificar novos genes ou mutações nos genes e facilmente projetar oligonucleotídeos baseados na sequência conhecida para experimentos adicionais.
O impacto da reação em cadeia da polimerase. Um dos problemas na clonagem molecular nos primeiros anos foi a obtenção de DNA inserido suficiente para clonar no vetor. Em 1983, Mullis desenvolveu uma técnica que resolveu este problema e revolucionou a clonagem molecular (28). Ele amplificou uma extensão do DNA alvo usando primers opostos para amplificar ambas as vertentes complementares do DNA, simultaneamente. Através de ciclos de desnaturação, recozimento e polimerização, ele mostrou que podia amplificar exponencialmente uma única cópia de DNA. A reacção em cadeia da polimerase, ou PCR, tornou possível amplificar e clonar genes a partir de quantidades previamente inadequadas de ADN. Para esta descoberta, Kary Mullis compartilhou o Prêmio Nobel de Química de 1993 “por contribuições ao desenvolvimento de métodos dentro da química baseada em DNA”.
Em 1970, Temin e Baltimore descobriram independentemente a transcriptase reversa em vírus, uma enzima que converte RNA em DNA (29,30). Logo após o desenvolvimento da PCR, a transcrição reversa foi acoplada à PCR (RT-PCR) para permitir a clonagem do RNA do mensageiro (mRNA). A transcrição reversa foi usada para criar uma cópia de DNA (cDNA) do mRNA que foi posteriormente amplificada por PCR para criar um inserto para ligadura. Por sua descoberta da enzima, Howard Temin e David Baltimore receberam o Prêmio Nobel de Medicina e Fisiologia de 1975, que compartilharam com Renato Dulbecco.
Clonagem de produtos PCR. O advento da PCR fez com que os pesquisadores pudessem agora clonar genes e segmentos de DNA com conhecimento limitado da seqüência amplicon. Entretanto, havia pouco consenso quanto ao método ideal de preparação de produtos PCR para uma ligação eficiente em vetores de clonagem.
Several diferentes métodos foram inicialmente usados para a clonagem de produtos PCR. O método mais simples, e ainda o mais comum, de clonagem de produtos PCR é através da introdução de locais de restrição nas extremidades do produto PCR (31). Isto permite a clonagem directa e direccional da inserção no vector após a digestão de restrição. A clonagem em bloco foi desenvolvida para ligar diretamente os produtos PCR gerados por polimerases que produziram pontas rombas, ou inserções projetadas para ter locais de restrição que deixaram pontas rombas uma vez que a inserção foi digerida. Isto foi útil na clonagem de fragmentos de DNA que não continham locais de restrição compatíveis com o vector (32).
Pouco depois da introdução da PCR, foi introduzida a PCR de extensão sobreposta como um método para montar produtos PCR numa sequência de DNA contígua (33). Neste método, a inserção de DNA é amplificada por PCR usando primers que geram um produto PCR contendo regiões sobrepostas com o vetor. O vetor e o insert são então misturados, desnaturados e recozidos, permitindo a hibridação do insert para o vetor. Uma segunda ronda de PCR gera moléculas de DNA recombinantes do vector contendo o inserto. A PCR de extensão sobreposta permitiu que os pesquisadores pudessem juntar grandes genes que não poderiam ser facilmente amplificados pelos métodos tradicionais de PCR. A PCR de extensão de sobreposição também foi utilizada para introduzir mutações em sequências de genes (34).
Figure 2. Visão geral da PCR

Desenvolvimento de técnicas especializadas de clonagem.
Num esforço para melhorar ainda mais a eficiência da clonagem molecular, foram desenvolvidas várias ferramentas e técnicas especializadas que exploraram as propriedades de enzimas únicas.
TA Clonagem. Uma abordagem aproveitou uma propriedade da Taq DNA Polimerase, a primeira polimerase termo-estável utilizada para PCR. Durante a amplificação, a Taq adiciona um único nucleótido de 3′ dA ao final de cada produto de PCR. O produto PCR pode ser facilmente ligado a um vector que foi cortado e concebido para conter um único resíduo T em cada filamento. Várias empresas comercializaram a técnica e vendem kits contendo vetores de clonagem que já são linearizados e “caudados”.
LIC. A clonagem independente de ligação (LIC), como seu nome indica, permite a união de moléculas de DNA na ausência de ligase de DNA. O LIC é comumente realizado com DNA Polimerase T4, que é utilizado para gerar sobreposições de DNA de cadeia única, >12 nucleotídeos longos, tanto no DNA vetorial linearizado quanto no inserto a ser clonado (35). Quando misturados, o vector e o inserto recozem através da longa extensão das extremidades compatíveis. O comprimento das extremidades compatíveis é suficiente para manter a molécula unida na ausência de ligase, mesmo durante a transformação. Uma vez transformadas, as lacunas são reparadas in vivo. Existem vários produtos comercialmente disponíveis para clonagem LIC.USER.
Clonagem USER foi desenvolvida pela primeira vez no início dos anos 90 como um método de clonagem independente de enzimas de restrição e ligas (36). Quando concebido pela primeira vez, o método baseou-se no uso de primers de PCR que continham uma cauda de 5′ de nucleotídeos ~12, em que pelo menos quatro bases de desoxitymidina tinham sido substituídas por desoxyuridinas. O produto PCR foi tratado com glicosidase de DNA uracil (UDG) e Endonuclease VIII, que excita as bases uracilares e deixa uma sobreposição de 3′ que pode ser recozida para um vector tratado de forma semelhante. A NEB vende a enzima USER para reacções de clonagem independentes de ligase e restrição enzimática.
Tendências Futuras
Clonagem molecular progrediu da clonagem de um único fragmento de ADN para a montagem de múltiplos componentes de ADN numa única estirpe contígua de ADN. Tecnologias novas e emergentes procuram transformar a clonagem num processo que é tão simples como organizar “blocos” de DNA uns ao lado dos outros.
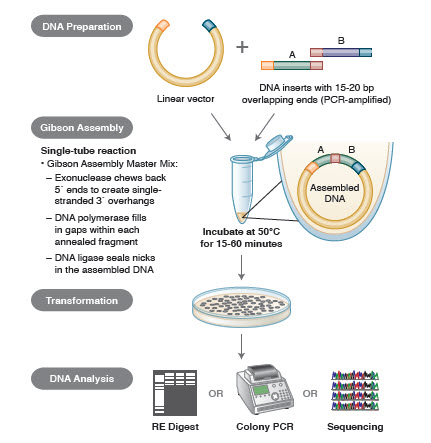
Métodos de montagem de ADN. Muitas tecnologias novas e elegantes permitem a montagem de múltiplos fragmentos de DNA em uma reação de um tubo. As vantagens dessas tecnologias são que elas são padronizadas, sem costura e na maioria das vezes independentes de seqüência. Além disso, a capacidade de montagem de múltiplos fragmentos de DNA em um tubo transforma uma série de reações de restrição/ligação anteriormente independentes em um procedimento aerodinâmico e eficiente.
Diferentes técnicas e produtos para a montagem de genes incluem SLIC (Sequence and Ligase Independent Cloning), Gibson Assembly (NEB), GeneArt® Seamless Cloning (Life Technologies) e Gateway® Cloning (Invitrogen) (35,37,38).
Na montagem de DNA, os blocos de DNA a serem montados são amplificados por PCR. Em seguida, os fragmentos de ADN a serem montados adjacentes uns aos outros são concebidos para conter blocos de sequências complementares que serão ligados entre si. Estas podem ser pontas coesas compatíveis, como as usadas para a Gibson Assembly, ou regiões contendo locais de reconhecimento para recombinações específicas do local (Gateway). A enzima usada para a ligação do DNA reconhecerá e reunirá cada conjunto de regiões compatíveis, criando uma única molécula de DNA contígua em uma reação.
Figure 3. Visão geral do Método de Clonagem do Conjunto Gibson
Biologia Sintética. A síntese de DNA é uma área da biologia sintética que atualmente está revolucionando a tecnologia do DNA recombinante. Embora um gene completo tenha sido sintetizado pela primeira vez in vitro em 1972 (40), a síntese de DNA de grandes moléculas de DNA só se tornou realidade no início dos anos 2000, quando os pesquisadores começaram a sintetizar genomas inteiros in vitro (41,42). Essas primeiras experiências levaram anos para serem concluídas, mas a tecnologia está acelerando a capacidade de sintetizar grandes moléculas de DNA.
Conclusão
Nos últimos 40 anos, a clonagem molecular progrediu do árduo isolamento e junção de dois pedaços de DNA, seguida de uma triagem intensiva de potenciais clones, para a montagem sem problemas de até 10 fragmentos de DNA com notável eficiência em apenas algumas horas, ou o desenho de moléculas de DNA em silico e sua sintetização in vitro. Juntas, todas essas tecnologias dão aos biólogos moleculares uma caixa de ferramentas surpreendentemente poderosa para explorar, manipular e aproveitar o DNA, o que ampliará ainda mais os horizontes da ciência. Entre as possibilidades estão o desenvolvimento de proteínas recombinantes mais seguras para o tratamento de doenças, o aperfeiçoamento da terapia genética (43) e a produção, validação e liberação mais rápida de novas vacinas (44). Mas, em última análise, o potencial é limitado apenas pela nossa imaginação.
Rebecca Tirabassi é um editor assistente em Bitesizebio.com.