Clonarea moleculară, un termen care a ajuns să însemne crearea de molecule de ADN recombinat, a stimulat progresul în toate științele vieții. Începând cu anii 1970, odată cu descoperirea endonucleazelor de restricție – enzime care taie în mod selectiv și specific moleculele de ADN – tehnologia ADN recombinant a cunoscut o creștere exponențială atât în ceea ce privește aplicațiile, cât și în ceea ce privește gradul de sofisticare, producând instrumente din ce în ce mai puternice pentru manipularea ADN-ului. Clonarea genelor este acum atât de simplă și eficientă încât a devenit o tehnică standard de laborator. Acest lucru a dus la o explozie a înțelegerii funcției genelor în ultimele decenii. Tehnologiile emergente promit posibilități și mai mari, cum ar fi posibilitatea cercetătorilor de a îmbina fără probleme mai multe fragmente de ADN și de a transforma plasmidele rezultate în bacterii, în mai puțin de două ore, sau utilizarea de casete de gene interschimbabile, care pot fi mutate cu ușurință între diferite construcții, pentru a maximiza viteza și flexibilitatea. În viitorul apropiat, clonarea moleculară va asista probabil la apariția unei noi paradigme, cu ajutorul tehnicilor de biologie sintetică, care vor permite sinteza chimică in vitro a oricărui construct de ADN specificat in silico. Aceste progrese ar trebui să permită construirea și iterația mai rapidă a clonelor de ADN, accelerând dezvoltarea vectorilor de terapie genică, a proceselor de producere a proteinelor recombinate și a noilor vaccinuri.
Rebecca Tirabassi, Bitesize Bio.
- Introducere
- Istoria clonării
- Fondamentele clonării moleculare
- Figura 1. Fluxul de lucru tradițional al clonării
- Consolidarea bazelor
- Figura 2. Prezentare generală a PCR
- Tendințe viitoare
- Figura 3. Prezentare generală a metodei de clonare Gibson Assembly
- Concluzie
- Vezi Ghidul nostru tehnic de clonare moleculară
Introducere
Clonarea moleculară se referă la izolarea unei secvențe de ADN din orice specie (adesea o genă) și inserția acesteia într-un vector pentru propagare, fără alterarea secvenței originale de ADN. Odată izolate, clonele moleculare pot fi utilizate pentru a genera multe copii ale ADN-ului pentru analiza secvenței genei și/sau pentru a exprima proteina rezultată în vederea studierii sau utilizării funcției proteinei. Clonele pot fi, de asemenea, manipulate și mutate in vitro pentru a modifica expresia și funcția proteinei.
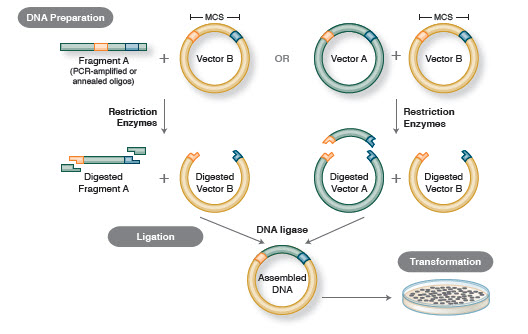
Fluxul de lucru de bază al clonării include patru etape:
- Isolarea fragmentelor de ADN țintă (adesea denumite inserții)
- Ligarea inserțiilor într-un vector de clonare adecvat, creând molecule recombinate (de ex, plasmide)
- Transformarea plasmidelor recombinante în bacterii sau în alte gazde adecvate pentru propagare
- Cercetarea/selecția gazdelor care conțin plasmidul recombinant dorit
Aceste patru etape revoluționare au fost asamblate cu atenție și realizate de mai multe laboratoare, începând cu sfârșitul anilor 1960 și începutul anilor 1970. Un rezumat al descoperirilor care cuprind clonarea moleculară tradițională este descris în paginile următoare.
Istoria clonării
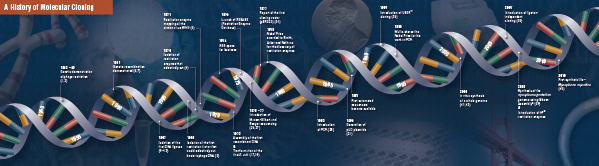
Fondamentele clonării moleculare
Tăierea (digestia). Tehnologia ADN recombinant a apărut pentru prima dată la sfârșitul anilor 1960, odată cu descoperirea enzimelor care puteau tăia și uni în mod specific moleculele de ADN bicatenar. De fapt, încă din 1952, două grupuri au observat independent că bacteriile codificau un „factor de restricție” care împiedica bacteriofagii să se dezvolte în anumite gazde (1,2). Dar natura factorului a fost descoperită abia în 1968, când Arber și Linn au reușit să izoleze o enzimă, denumită factor de restricție, care tăia selectiv ADN exogen, dar nu și ADN bacterian (3). Aceste studii au identificat, de asemenea, o enzimă metilază care proteja ADN-ul bacterian de enzimele de restricție.
La scurt timp după descoperirea lui Arber și Linn, Smith a extins și confirmat aceste studii prin izolarea unei enzime de restricție din Haemophilus influenza. El a demonstrat că enzima taie selectiv ADN-ul în mijlocul unei secvențe specifice de 6 perechi de baze de ADN; o caracteristică a anumitor enzime de restricție este propensiunea lor de a tăia substratul ADN în sau în apropierea unor secvențe de „recunoaștere” specifice, adesea palindromice (4).
Puterea deplină a enzimelor de restricție nu a fost conștientizată până când enzimele de restricție și electroforeza pe gel au fost folosite pentru a cartografia genomul virusului Simian 40 (SV40) (5). Pentru aceste descoperiri fundamentale, Werner Arber, Hamilton Smith și Daniel Nathans au împărțit Premiul Nobel pentru Medicină în 1978.
Figura 1. Fluxul de lucru tradițional al clonării
Assembling (Ligation). La fel ca și descoperirea enzimelor care taie ADN-ul, descoperirea unei enzime care poate uni ADN-ul a fost precedată de observații anterioare, proeminente. La începutul anilor 1960, două grupuri au descoperit că recombinarea genetică ar putea avea loc prin ruperea și legarea moleculelor de ADN (6,7), urmată îndeaproape de observația că ADN-ul bacteriofagului liniar este transformat rapid în cercuri închise covalent după infectarea gazdei (8). Doar doi ani mai târziu, cinci grupuri au izolat independent ADN-ligaze și au demonstrat capacitatea acestora de a asambla două bucăți de ADN (9-13).
La scurt timp după descoperirea enzimelor de restricție și a ligazelor ADN, a fost realizată prima moleculă de ADN recombinat. În 1972, Berg a tăiat și a legat separat o bucată de ADN al bacteriofagului lambda sau al operonului galactozei din E. coli cu ADN SV40 pentru a crea primele molecule de ADN recombinant (14). Aceste studii au deschis calea conceptului că, datorită naturii universale a ADN-ului, ADN-ul din orice specie poate fi unit. În 1980, Paul Berg a împărțit Premiul Nobel pentru Chimie cu Walter Gilbert și Frederick Sanger (cei care au dezvoltat secvențierea ADN-ului), pentru „studiile sale fundamentale privind biochimia acizilor nucleici, în special în ceea ce privește ADN-ul recombinant”.
Transformare. Tehnologia ADN recombinant ar fi foarte limitată, iar clonarea moleculară ar fi imposibilă, fără mijloacele de propagare și izolare a moleculei de ADN nou construite. Capacitatea de a transforma bacteriile, sau de a induce absorbția, încorporarea și exprimarea materialului genetic străin, a fost demonstrată pentru prima dată de Griffith atunci când a transformat o tulpină neletală de bacterii într-o tulpină letală prin amestecarea tulpinii neletale cu bacterii letale inactivate termic (15). Cu toate acestea, natura „principiului de transformare” care a transmis letalitatea nu a fost înțeleasă până în 1944. În același an, Avery, Macleod și McCarty au demonstrat că ADN-ul, și nu proteinele, era responsabil pentru inducerea fenotipului letal (16).
Început, s-a crezut că tulpina bacteriană obișnuită de laborator, E. coli, era refractară la transformare, până când Mandel și Higa au demonstrat că tratarea E. coli cu clorură de calciu a indus absorbția ADN-ului bacteriofag (17). Cohen a aplicat acest principiu, în 1972, când a fost pionierul transformării bacteriilor cu plasmide pentru a le conferi bacteriilor rezistență la antibiotice (18).
Experimentul suprem: digestia, ligaturarea și transformarea unei molecule de ADN recombinant a fost executat de Boyer, Cohen și Chang în 1973, când au digerat plasmidul pSC101 cu EcoRI, au ligaturat fragmentul liniarizat la un alt plasmid restricționat enzimatic și au transformat molecula recombinantă rezultată în E. coli, conferind bacteriei rezistență la tetraciclină (19), punând astfel bazele majorității lucrărilor de ADN recombinant de atunci.
Consolidarea bazelor
În timp ce oamenii de știință descoperiseră și aplicaseră toate principiile de bază pentru crearea și propagarea ADN recombinant în bacterii, procesul era ineficient. Preparatele de enzime de restricție erau nesigure din cauza procedurilor de purificare nestandardizate, plasmidele pentru clonare erau greoaie, greu de lucrat și limitate ca număr, iar experimentele erau limitate de cantitatea de ADN inserat care putea fi izolată. Cercetările din următoarele câteva decenii au dus la îmbunătățirea tehnicilor și instrumentelor disponibile pentru clonarea moleculară.
Proiectarea timpurie a vectorilor.
Dezvoltarea primului vector standardizat. Oamenii de știință care lucrau în laboratorul lui Boyer au recunoscut necesitatea unei plasmide de clonare generală, o plasmidă compactă cu situsuri de restricție unice pentru clonarea în ADN străin și exprimarea genelor de rezistență la antibiotice pentru selectarea bacteriilor transformate. În 1977, aceștia au descris primul vector conceput în scopul clonării, pBR322 (20). Acest vector era mic, cu o dimensiune de ~4 kilobaze, și avea două gene de rezistență la antibiotice pentru selecție.
Vectori cu selecție la bord și randamente mai mari. Deși selecția cu antibiotice a împiedicat creșterea bacteriilor netransformate, plasmidele care se relegau fără fragmente de ADN inserat (autolegare) puteau totuși conferi bacteriilor rezistență la antibiotice. Prin urmare, găsirea clonelor bacteriene corecte care conțin molecula de ADN recombinant dorită ar putea fi consumatoare de timp.
Vieira și Messing au conceput un instrument de screening pentru a identifica coloniile bacteriene care conțin plasmide cu inserții de ADN. Pe baza plasmidului pBR322, ei au creat seria de plasmide pUC, care conținea un sistem de „screening albastru/alb” (21). Plasarea unui situs de clonare multiplă (MCS) care conține mai multe situsuri de restricție unice în cadrul genei LacZ´ a permis cercetătorilor să depisteze coloniile bacteriene care conțin plasmide cu inserția de ADN străin. Atunci când bacteriile au fost plasate pe mediile corecte, coloniile albe conțineau plasmide cu inserții, în timp ce coloniile albastre conțineau plasmide fără inserții. Plasmidele pUC aveau un avantaj suplimentar față de vectorii existenți; ele conțineau o mutație care a dus la un număr mai mare de copii, crescând astfel randamentul plasmidelor.
Îmbunătățirea digestărilor de restricție. Primele lucrări cu enzime de restricție au fost împiedicate de puritatea preparatului enzimatic și de lipsa de înțelegere a cerințelor de tampon pentru fiecare enzimă. În 1975, New England Biolabs (NEB) a devenit prima companie care a comercializat enzime de restricție produse dintr-o sursă recombinantă. Acest lucru a permis obținerea unor randamente mai mari, o puritate îmbunătățită, consistență de la lot la lot și prețuri mai mici. În prezent, peste 4.000 de enzime de restricție, care recunosc peste 300 de secvențe diferite, au fost descoperite de oamenii de știință din întreaga lume . NEB furnizează în prezent peste 230 dintre aceste specificități.
NEB a fost, de asemenea, una dintre primele companii care a dezvoltat un sistem standardizat cu patru tampoane și care a caracterizat toate activitățile enzimelor sale în acest sistem tampon. Acest lucru a dus la o mai bună înțelegere a modului în care se poate realiza o dublă digestie, sau digestia ADN-ului cu două enzime în mod sim-ultanat. Cercetările ulterioare au dus la dezvoltarea sistemelor cu un singur tampon, care sunt compatibile cu cele mai comune enzime de restricție (cum ar fi Tamponul CutSmart™ de la NEB).
Cu apariția bibliotecilor de enzime de restricție disponibile în comerț cu specificități de secvență cunoscute, enzimele de restricție au devenit un instrument puternic pentru depistarea potențialelor clone de ADN recombinant. „Digestia de diagnosticare” a fost și este încă una dintre cele mai comune tehnici utilizate în clonarea moleculară.
Prepararea vectorilor și a inserțiilor. Eficiența și versatilitatea clonării au fost, de asemenea, îmbunătățite prin dezvoltarea diferitelor tehnici de pregătire a vectorilor înainte de ligaturare. Au fost izolate fosfataze alcaline care puteau îndepărta grupările fosfat 3´ și 5´ de la capetele ADN-ului . În scurt timp s-a descoperit că tratarea vectorilor cu fosfatază intestinală de vițel (CIP) defosforila capetele ADN-ului și împiedica autoligarea vectorului, crescând recuperarea plasmidelor cu inserție (24).
Enzima CIP s-a dovedit a fi dificil de inactivat, iar orice activitate reziduală a dus la defosforilarea ADN-ului inserat și la inhibarea reacției de ligare. Descoperirea fosfatazelor alcaline termolabile, cum ar fi fosfataza alcalină recombinantă Shrimp Alkaline Phosphatase (rSAP) și Antarctic Phosphatase (AP) (ambele comercializate de NEB), a redus etapele și timpul necesar, deoarece o simplă schimbare de temperatură inactivează enzima înainte de etapa de ligare (25).
Sosește secvențierea ADN. Secvențierea ADN a fost dezvoltată la sfârșitul anilor 1970, când au fost concepute două metode concurente. Maxam și Gilbert au dezvoltat „metoda de secvențiere chimică”, care se baza pe modificarea chimică a ADN-ului și pe scindarea ulterioară la baze specifice (26). În același timp, Sanger și colegii săi au publicat „metoda de terminare a lanțului”, care a devenit metoda utilizată de majoritatea cercetătorilor (27). Metoda Sanger a devenit rapid automatizată, iar primele secvențiatoare automate au fost vândute în 1987.
Capacitatea de a determina secvența unei porțiuni de ADN a sporit fiabilitatea și versatilitatea clonării moleculare. Odată clonate, oamenii de știință puteau să secvențieze clonele pentru a identifica definitiv molecula recombinantă corectă, să identifice noi gene sau mutații în gene și să proiecteze cu ușurință oligonucleotide pe baza secvenței cunoscute pentru experimente suplimentare.
Impact al reacției în lanț a polimerazei. Una dintre problemele clonării moleculare din primii ani a fost obținerea unei cantități suficiente de ADN inserat pentru a fi clonat în vector. În 1983, Mullis a conceput o tehnică care a rezolvat această problemă și a revoluționat clonarea moleculară (28). El a amplificat o porțiune de ADN țintă prin utilizarea unor primeri opuși pentru a amplifica ambele șiruri complementare de ADN, simultan. Prin cicluri de denaturare, recoacere și polimerizare, el a demonstrat că poate amplifica exponențial o singură copie de ADN. Reacția în lanț a polimerazei, sau PCR, a făcut posibilă amplificarea și clonarea genelor din cantități de ADN anterior inadecvate. Pentru această descoperire, Kary Mullis a împărțit Premiul Nobel pentru Chimie din 1993 „pentru contribuții la dezvoltarea metodelor din cadrul chimiei bazate pe ADN”.
În 1970, Temin și Baltimore au descoperit independent transcriptaza inversă în virusuri, o enzimă care transformă ARN-ul în ADN (29,30). La scurt timp după dezvoltarea PCR, transcrierea inversă a fost cuplată cu PCR (RT-PCR) pentru a permite clonarea ARN-ului mesager (ARNm). Transcrierea inversă a fost utilizată pentru a crea o copie ADN (ADNc) a ARNm care a fost ulterior amplificată prin PCR pentru a crea o inserție pentru ligare. Pentru descoperirea enzimei, Howard Temin și David Baltimore au primit în 1975 Premiul Nobel pentru Medicină și Fiziologie, pe care l-au împărțit cu Renato Dulbecco.
Clonarea produselor PCR. Apariția PCR a însemnat că cercetătorii puteau acum să cloneze gene și segmente de ADN cu cunoștințe limitate despre secvența ampliconului. Cu toate acestea, nu a existat prea mult consens în ceea ce privește metoda optimă de preparare a produselor PCR pentru o ligare eficientă în vectorii de clonare.
La început au fost folosite mai multe metode diferite pentru clonarea produselor PCR. Cea mai simplă și încă cea mai frecventă metodă de clonare a produselor PCR este prin introducerea de situsuri de restricție la capetele produsului PCR (31). Acest lucru permite clonarea directă și direcțională a inserției în vector după digestia de restricție. Clonarea cu capetele blunt-ended a fost dezvoltată pentru a lega direct produsele PCR generate de polimeraze care au produs capete blunt-ended sau inserții modificate pentru a avea situsuri de restricție care au lăsat capetele blunt-ended după ce inserția a fost digerată. Acest lucru a fost util în clonarea fragmentelor de ADN care nu conțineau situsuri de restricție compatibile cu vectorul (32).
La scurt timp după introducerea PCR, a fost introdusă PCR cu extensie de suprapunere ca metodă de asamblare a produselor PCR într-o singură secvență de ADN contiguă (33). În această metodă, insertul de ADN este amplificat prin PCR folosind primeri care generează un produs PCR care conține regiuni suprapuse cu vectorul. Vectorul și insertul sunt apoi amestecate, denaturate și aneantizate, permițând hibridizarea insertului cu vectorul. O a doua rundă de PCR generează molecule de ADN recombinant de vector care conține inserția. PCR cu extensie de suprapunere a permis cercetătorilor să pună cap la cap gene mari care nu puteau fi amplificate cu ușurință prin metodele PCR tradiționale. Overlap extension PCR a fost, de asemenea, utilizată pentru a introduce mutații în secvențe de gene (34).
Figura 2. Prezentare generală a PCR

Dezvoltarea tehnicilor specializate de clonare.
În efortul de a îmbunătăți și mai mult eficiența clonării moleculare, au fost dezvoltate mai multe instrumente și tehnici specializate care au exploatat proprietățile unor enzime unice.
Clonarea TA. O abordare a profitat de o proprietate a Taq ADN Polimerazei Taq, prima polimerază stabilă la căldură utilizată pentru PCR. În timpul amplificării, Taq adaugă o singură nucleotidă 3´ dA la capătul fiecărui produs PCR. Produsul PCR poate fi ușor ligat într-un vector care a fost tăiat și modificat pentru a conține reziduuri T unice pe fiecare catenă. Mai multe companii au comercializat această tehnică și vând kituri care conțin vectori de clonare care sunt deja liniarizați și cu „coadă”.
LIC. Clonarea independentă de ligaturare (LIC), după cum îi spune și numele, permite îmbinarea moleculelor de ADN în absența ADN ligazei. LIC se realizează în mod obișnuit cu ADN polimeraza T4 ADN, care este utilizată pentru a genera supraînălțări de ADN monocatenar, cu o lungime de >12 nucleotide, atât pe ADN-ul vector liniarizat, cât și pe inserția care urmează să fie clonată (35). Atunci când sunt amestecate, vectorul și insertul se annează prin întinderea lungă a capetelor compatibile. Lungimea capetelor compatibile este suficientă pentru a menține molecula împreună în absența ligazei, chiar și în timpul transformării. Odată transformate, lacunele sunt reparate in vivo. Există mai multe produse diferite disponibile în comerț pentru clonarea LIC.USER.
Clonarea USER a fost dezvoltată pentru prima dată la începutul anilor 1990 ca metodă de clonare independentă de enzimele de restricție și de ligaze (36). Când a fost concepută pentru prima dată, metoda se baza pe utilizarea de primeri PCR care conțineau o coadă 5´ de ~12 nucleotide, în care cel puțin patru baze deoxitimidinice fuseseră înlocuite cu deoxiuridine. Produsul PCR a fost tratat cu ADN-glicozidază uracil ADN (UDG) și Endonucleasa VIII, care extirpă bazele uracil și lasă o suprapunere 3´ care poate fi aneantizată la un vector tratat în mod similar. NEB vinde enzima USER pentru reacții de clonare independente de ligazele și enzimele de restricție.
Tendințe viitoare
Clonarea moleculară a progresat de la clonarea unui singur fragment de ADN la asamblarea mai multor componente de ADN într-o singură porțiune contiguă de ADN. Tehnologiile noi și emergente urmăresc să transforme clonarea într-un proces la fel de simplu ca și aranjarea unor „blocuri” de ADN unul lângă altul.
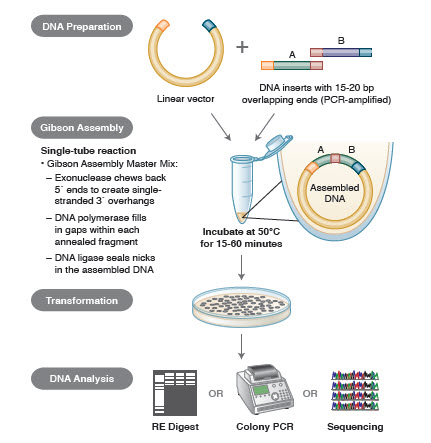
Metode de asamblare a ADN. Multe tehnologii noi și elegante permit asamblarea mai multor fragmente de ADN într-o singură reacție tubulară. Avantajele acestor tehnologii sunt că sunt standardizate, fără întreruperi și în mare parte independente de secvență. În plus, capacitatea de a asambla mai multe fragmente de ADN într-un singur tub transformă o serie de reacții de restricție/ligare, anterior independente, într-o procedură raționalizată și eficientă.
Diferite tehnici și produse pentru asamblarea genelor includ SLIC (Sequence and Ligase Independent Cloning), Gibson Assembly (NEB), GeneArt® Seamless Cloning (Life Technologies) și Gateway® Cloning (Invitrogen) (35,37,38).
În asamblarea ADN, blocurile de ADN care urmează să fie asamblate sunt amplificate prin PCR. Apoi, fragmentele de ADN care urmează să fie asamblate adiacente unul de altul sunt proiectate pentru a conține blocuri de secvențe complementare care vor fi ligaturate împreună. Acestea ar putea fi capete de coeziune compatibile, cum ar fi cele utilizate pentru asamblarea Gibson, sau regiuni care conțin situsuri de recunoaștere pentru recombinazele site-specifice (Gateway). Enzima utilizată pentru ligatura ADN va recunoaște și asambla fiecare set de regiuni compatibile, creând o singură moleculă de ADN contiguă într-o singură reacție.
Figura 3. Prezentare generală a metodei de clonare Gibson Assembly
Biologie sintetică. Sinteza ADN este un domeniu al biologiei sintetice care revoluționează în prezent tehnologia ADN recombinant. Deși o genă completă a fost sintetizată pentru prima dată in vitro în 1972 (40), sinteza ADN a moleculelor mari de ADN nu a devenit o realitate până la începutul anilor 2000, când cercetătorii au început să sintetizeze genomuri întregi in vitro (41,42). Aceste experimente timpurii au avut nevoie de ani pentru a fi finalizate, dar tehnologia accelerează capacitatea de a sintetiza molecule mari de ADN.
Concluzie
În ultimii 40 de ani, clonarea moleculară a progresat de la izolarea și asamblarea dificilă a două bucăți de ADN, urmată de un screening intensiv al potențialelor clone, la asamblarea fără probleme a până la 10 fragmente de ADN cu o eficiență remarcabilă în doar câteva ore, sau proiectarea moleculelor de ADN in silico și sintetizarea lor in vitro. Împreună, toate aceste tehnologii oferă biologilor moleculari un set de instrumente uimitor de puternice pentru explorarea, manipularea și valorificarea ADN-ului, care vor lărgi și mai mult orizonturile științei. Printre posibilități se numără dezvoltarea unor proteine recombinante mai sigure pentru tratamentul bolilor, îmbunătățirea terapiei genice (43) și producerea, validarea și eliberarea mai rapidă a noilor vaccinuri (44). Dar, în cele din urmă, potențialul este constrâns doar de imaginația noastră.
Rebecca Tirabassi este redactor asistent la Bitesizebio.com.
Vezi Ghidul nostru tehnic de clonare moleculară
.